Nicholas Ingolia: Barcoding the Genome
By Kirsten Mickelwait
 Nicholas Ingolia, MCB associate professor of biochemistry, biophysics and structural biology. |
When COVID-19 caused the world to pause and the Bay Area to go into “lockdown,” scientific labs across the Berkeley campus nearly ground to a halt. The lab of Nicholas Ingolia, associate professor of molecular and cell biology, was among them. But after being out of the lab for nearly three months, his team gradually ramped up again. “I’ve been impressed by how conscientious my group has been, so we can share time in the lab in a way that keeps everyone safe while moving their science forward,” he says. What’s more, because some of the lab’s research is RNA-focused, and because SARS-CoV-2 is an RNA virus, the team has been able to collaborate with other Berkeley researchers to study RNA biology on the virus.
The Ingolia lab has also developed a new technique that will make it easier to map genetic networks. The research team studies translational control of gene expression through global and genome-wide analyses; using the CRISPR-Cas9 technology, it’s discovered a new way to manipulate genes individually and rapidly see the impact across the entire genetic network. The technique is called CiBER-seq: CRISPR interference with Barcoded Expression Reporter sequencing. It allows researchers to perform thousands of experiments at once, quickly identifying the regulatory sequences that control disease genes and potentially helping to find new targets for drugs.
|
|
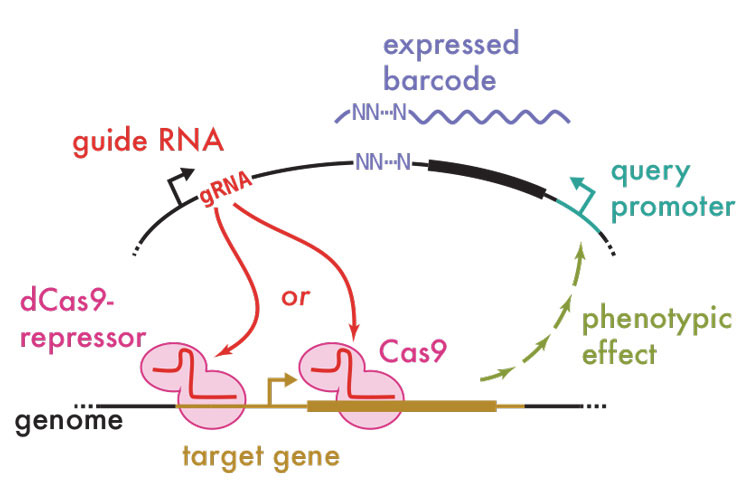
“With CRISPR, it’s now possible to systematically disrupt each gene in the genome one at a time, in a very programmable way,” says Ingolia. “This is really powerful as a research tool. If we can perturb each gene one by one, how can we measure the changes that each of those genetic perturbations provokes in the cell?”
While many scientists work “downstream,” examining effects that occur after gene manipulation, Ingolia’s lab works “upstream” to learn what causes those effects. The approach has many potential applications in disease research. “Maybe we know that some gene is switched on in a disease, and we want to know what happens upstream that causes that gene to be switched on,” Ingolia explains. “Our approach allows us to ask that question in a very direct way. So we can systemically ask, for a certain gene, what are all the genetic changes that will switch it on or off?”
 Ingolia lab in 2019 in front of Li Ka Shing building. |
The group’s work on CiBER-seq was published in Science in December 2020, after lead author Ryan Muller, a graduate student in Ingolia’s lab, conceived of the initial idea in June 2018. “One Friday afternoon, just a few hours before I had to catch a flight, Nick agreed to a last-minute meeting to discuss what would become the basis of CiBER-seq,” Muller says. “His willingness to consider my hastily drawn scribbles, and the exchange of improvements and applications he suggested, epitomizes both Nick's scientific vision and the kind of mentor he strives to be. Throughout the course of my graduate career, he’s been there for me as an inspiring example of thoughtful science, clever approaches, and supportive mentorship.”
For an in-depth look at their technique to tease apart genetic networks, read these articles in Berkeley News and Oncology Times. Find out more about the Ingolia lab and their research here.
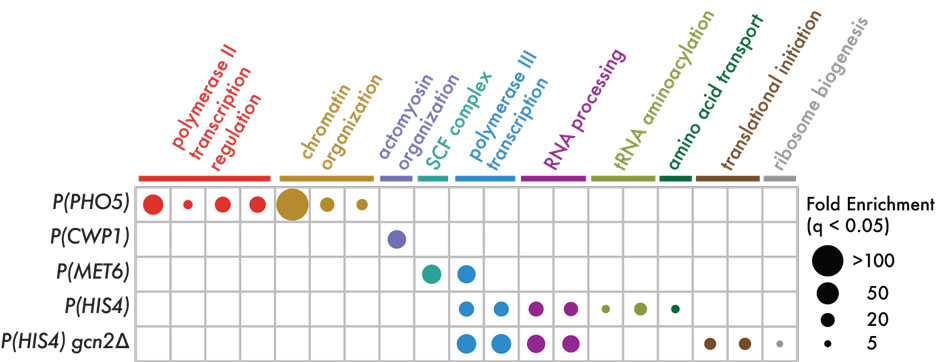 |
| Excerpt from Fig. 1 of work published in Science: Barcoded expression reporters linked transcriptional responses with gRNA-mediated perturbations in massively parallel screens. Pictured here is part (F) GO analysis of each CiBER-seq query promoter profile. Guides were filtered by q < 0.05 and >2-fold increase, and resulting gene lists were analyzed for GO category overrepresentation using Fisher’s exact test with false discovery rate (FDR)–adjusted P < 0.05. The most statistically significant entry was chosen from chains of hierarchically nested categories, and all chains with significant categories for any promoter are represented in the plot. |
Back to Main Spring 2021 Newsletter Page
| Connect With Us! | ||||
MCB Twitter |
 MCB Facebook |
 LinkedIn Postdocs, PhDs, or Undergrads |
 Cal Alumni Network |
 Give to MCB |
