Barbara Meyer

Howard Hughes Medical Institute Investigator, Professor of Genetics, Genomics, Evolution, and Development
Lab Homepage: http://mcb.berkeley.edu/labs/meyerResearch Interests
Current Projects
Counting Chromosomes to Determine Sex: Molecular Antagonism between X-Chromosome, Autosome Signals Specifies Nematode Sex
Many organisms determine sexual fate by a chromosome-counting mechanism that distinguishes one X chromosome from two. Embryos with one X become males, while those with two become females. We dissected the molecular mechanism by which the nematode C. elegans counts its sex chromosomes to discern how small changes in the concentrations of molecular signals are translated into dramatically different developmental fates. C. elegans tallies X-chromosome number relative to the ploidy, the sets of autosomes (X:A signal). It discriminates with high fidelity between tiny differences in the signal: 2X:3A embryos (ratio 0.67) become males, while 3X:4A embryos (ratio 0.75) become hermaphrodites (Figure 1).
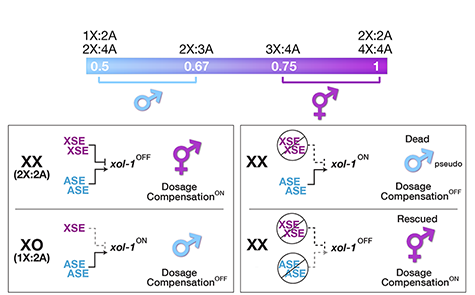
Figure 1. The X:A sex determining signal.
We showed that a set of X-linked genes called X-signal elements (XSEs) communicates X-chromosome dose by repressing the master sex-determination switch gene xol-1 in a cumulative, dose-dependent manner. XOL-1, a GHMP kinase, is activated in 1X:2A embryos (1 dose of XSEs) to set the male fate but repressed in 2X:2A embryos (2 doses of XSEs) to promote the hermaphrodite fate, including the activation of X-chromosome dosage compensation. We also showed that the dose of autosomes is communicated by a set of autosomal signal elements (ASEs) that also act in a cumulative, dose-dependent manner to counter XSEs by stimulating xol-1 transcription. We have explored the biochemical basis by which XSEs counter ASEs to determine sex. Analysis in vitro showed that XSEs (nuclear receptors and homeodomain proteins) and ASEs (T-box and zinc-finger proteins) bind directly to at least 5 distinct sites in xol-1 regulatory DNA to counteract each other's activities and thereby regulate xol-1 transcription (Figure 2). Analysis in vivo showed that disrupting ASE and XSE binding sites recapitulated the mis-regulation of xol-1 transcription caused by disrupting the cognate signal element genes. XSE and ASE binding sites are distinct and non-overlapping, suggesting that direct competition for xol-1 binding is not the mechanism by which XSEs counter ASEs. Instead, XSEs likely antagonize ASEs by recruiting cofactors with reciprocal activities that induce opposite transcriptional states. The X:A balance is thus communicated in part through multiple antagonistic molecular interactions carried out on a single promoter, revealing how small differences in X:A values can elicit different sexual fates. We are currently identifying potential coactivators and corepressors and directing efforts toward understanding the evolution of the X:A signal across nematode species.
Although most XSEs repress xol-1 by regulating transcription, one XSE, an RNA binding protein, represses xol-1 by binding to an alternatively spliced intron and blocking its proper splicing, thereby generating a non-functional transcript with an in-frame stop codon (Figure 2). This second tier of repression enhances the fidelity of the counting process.
The concept of a sex signal comprising competing XSEs and ASEs arose as a theory for fruit flies one century ago, and it subsequently became entrenched in textbooks. Ironically, the recent work of others showed the fly sex signal does not fit this simple paradigm, but our work shows the worm signal does.
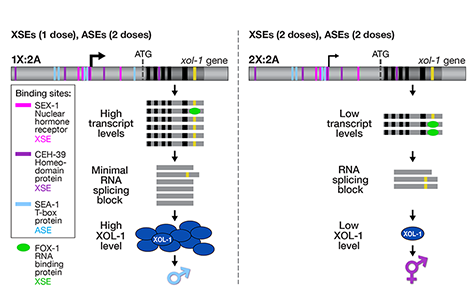
Figure 2. Model depicting the molecular antagonism between XSEs and ASEs that determines sex.
X-Chromosome Dosage Compensation: Repressing X Chromosomes via Molecular Machines.
Organisms that use sex chromosomes to determine sexual fate evolved the essential, chromosome-wide regulatory process called dosage compensation to balance X-chromosome gene expression between the sexes. Strategies for dosage compensation differ from worms to mammals, but invariably a regulatory complex is targeted to X chromosomes of one sex to modulate transcription along the entire chromosome. The heritable, regulation of X-chromosome expression during dosage compensation is exemplary for dissecting the coordinate regulation of gene expression over large chromosomal territories and the role of chromosome structure in regulating gene expression.
We defined the C. elegans dosage compensation complex (DCC) and showed it is homologous to condensin, a conserved protein complex that mediates the compaction, resolution, and segregation of mitotic and meiotic chromosomes from yeast to humans (Figure 3). The DCC binds to both X chromosomes of hermaphrodites to reduce transcription by half (Figure 3). Failure to reduce expression kills hermaphrodites. Most DCC condensin subunits also control the structure and function of mitotic and meiotic chromosomes by participating in two other distinct condensin complexes (Figure 3). Not only has the DCC co-opt subunits of condensin to control gene expression, it co-opted a subunit from the MLL/COMPASS complex, a histone modifying complex, to help recruit condensin subunits to rex sites.
We found that the DCC condensin subunits are recruited specifically to hermaphrodite X chromosomes by sex-specific DCC subunits that trigger binding to cis-acting regulatory elements on X, called rex and dox sites. rex (recruitment elements on X) sites recruit the DCC in an autonomous, sequence-dependent manner using DNA motifs highly enriched on X chromosomes. The DCC spreads to dox (dependent on X) sites, which reside in promoters of active genes and bind the DCC robustly only when linked to rex sites.
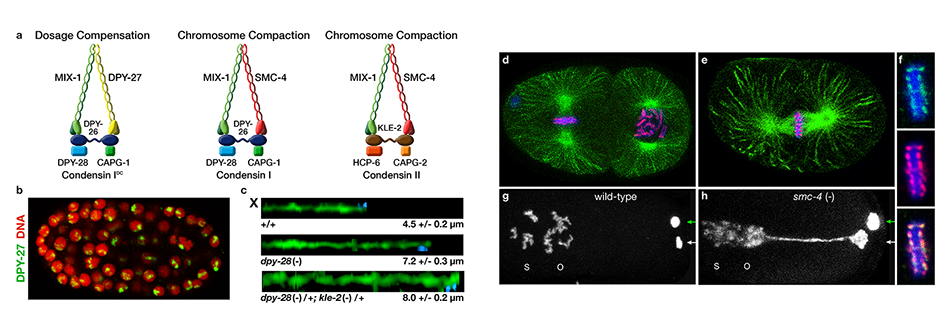
Figure 3. Biochemically distinct condensin complexes with interchangeable subunits control chromosome
structure throughout C. elegans development.
Dynamic Control of X-Chromosome Conformation and Repression by a Histone H4K20me Demethylase.
We recently found that DCC subunit DPY-21 has a histone demethylase activity that is responsible for the selective enrichment of H4K20me1 on X chromosomes of XX embryos upon DCC binding. X-ray crystallography and biochemical assays of DPY-21 revealed a novel subfamily of Jumonji C histone demethylases that converts H4K20me2 to H4K20me1. Selective inactivation of demethylase activity in vivo by genome editing eliminated H4K20me1 enrichment on X, elevated X-linked gene expression, reduced X-chromosome compaction, and disrupted X-chromosome topology by weakening TAD boundaries. These findings, among others, demonstrate the direct impact of chromatin modification on higher-order chromosome structure in long-range regulation of gene expression.
H4K20me1 is also enriched on the mammalian inactive X chromosome, but the role of this enrichment in mammalian transcriptional silencing is not known, nor is a selective reagent available to test its role. We showed that the mouse homolog of the DCC subunit also has H4K20me2 demethylase activity. Hence the worm system holds great promise for understanding the effects of histone modification in mammals.
Unexpectedly, DPY-21 associates with autosomes but not X chromosomes of germ cells in a DCC-independent manner to enrich H4K20me1 and facilitate chromosome compaction. Thus, DPY-21 is an adaptable chromatin regulator that is harnessed during development for distinct biological functions. In both somatic cells and germ cells, H4K20me1 enrichment modulates 3D chromosome architecture to carry out these functions (Figure 4).
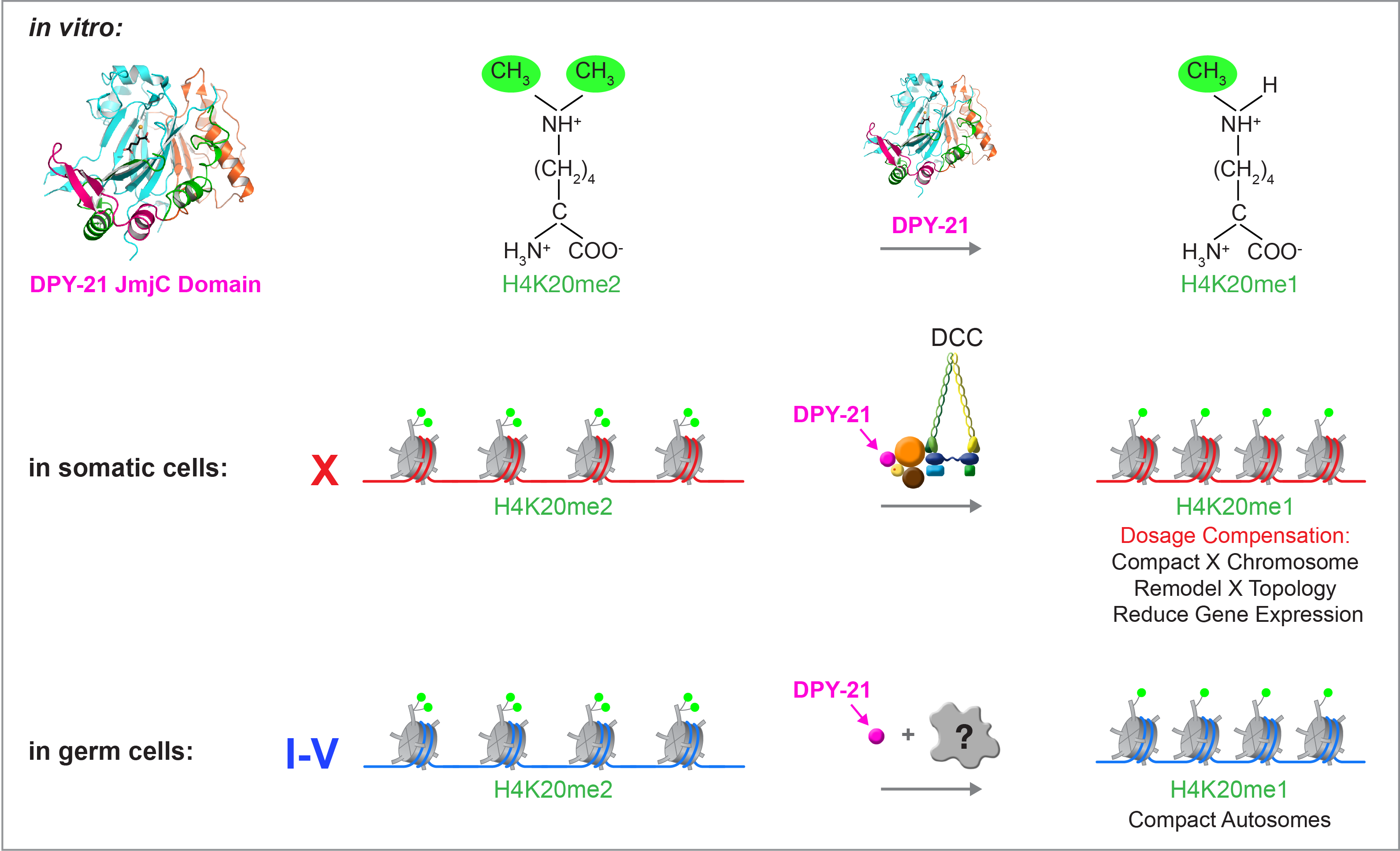
Figure 4. DPY-21 is a chromatin regulator that is harnessed during development for different biological functions. DPY-21 crystal structure and biochemical activity revealed a novel H4K20me2 Jumonji C demethylase. In somatic cells, DPY-21 enriches H4K20me1 on X chromosomes to repress gene expression. H4K20me1 enrichment controls the higher-order structure of X chromosomes. In germ cells, DPY-21 enriches H4K20me1 on autosomes in a DCC-independent manner to compact autosomes.
Condensin-driven remodeling of X-chromosome topology during dosage compensation.
The three-dimensional organization of a genome plays a critical role in regulating gene expression, yet little is known about the machinery and mechanisms that determine higher-order chromosome structure. The involvement of bona fide condensin subunits in dosage compensation together with our observation that the DCC acts at a distance to regulate gene expression suggested that the DCC might alter the topology of X chromosomes to reduce gene expression chromosome wide.
Using genome-wide chromosome conformation capture techniques (in collaboration with Job Dekker's lab at U. Mass. Worcester) with single-cell fluorescence in situ hybridization and RNA-seq to compare chromosome structure and gene expression in wild-type and dosage-compensation-defective embryos, we showed that the DCC remodels X chromosomes of hermaphrodites into a unique, sex-specific spatial conformation, distinct from autosomes, using its highest-affinity rex sites to facilitate long-range interactions across X. Dosage-compensated X chromosomes consist of self-interacting domains (~ 1 Mb) resembling mammalian Topologically Associating Domains (TADs). TADs on X have stronger boundaries and more regular spacing than those on autosomes. Many TAD boundaries on X coincide with the highest-affinity rex sites, and these boundaries become diminished or lost in mutants lacking DCC binding, causing the structure of X to resemble that of autosomes. These results predicted that deletion of an endogenous rex site at a DCC-dependent boundary should disrupt the boundary. As predicted, Cas9-mediated deletion of a rex site greatly diminished the boundary, further demonstrating the condensin-driven remodeling of X-chromosome topology during dosage compensation. Thus, condensin acts as a key structural element to reorganize interphase chromosomes and thereby regulate gene expression. Prior to our work, no molecular trigger or set of DNA binding sites was known to cause a comparably strong effect on TAD structure in higher eukaryotes. Our understanding of the topology of dosage-compensated X chromosomes provides fertile ground to decipher the detailed mechanistic relationship between higher-order chromosome structure and chromosome-wide regulation of gene expression (Figure 5).
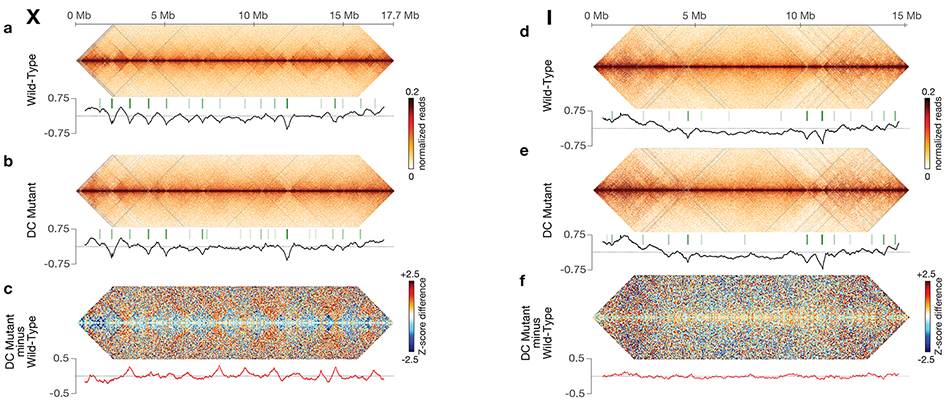
Figure 5. DCC modulates spatial organization of X chromosomes.
Selected Publications
Farboud, B., Severson, A.F., Meyer, B.J. (2018). Strategies for Efficient Genome Editing Using CRISPR-Cas9. Genetics. DOI: 10.1534/genetics.118.301775 PMID 30504364
Yin, D., Schwarz, E.M., Thomas, C.G., Felde, R.L., Korf, I.F., Cutter, A.D., Schartner, C.M., Ralston, E.J., Meyer, B.J., Haag, E.S. (2018). Rapid Genome Shrinkage in a Self-Fertile Nematode Reveals Sperm Competition Proteins. Science 359: 55-61. DOI: 10.1126/science.aao0827 PMID: 29302007 PMCID: PMC5789457
Brejc, K., Bian, Q., Uzawa, S., Wheeler, B.S., Anderson, E.C., King, D.S., Kranzusch, P.J., Preston, C.G., Meyer, B.J. (2017). Dynamic Control of X-Chromosome Conformation and Repression by a Histone H4K20 Demethylase. Cell 171: 85-102.e23. doi: 10.1016/j.cell.2017.07.041. Epub 2017 Aug 31. PMID: 28867287 PMCID: PMC5678999
Wheeler, B.S., Anderson, E., Frøkjær-Jensen, C., Bian, Q., Jorgensen, E., Meyer, B.J. (2016). Chromosome-Wide Mechanisms to Decouple Gene Expression from Gene Dose during Sex-Chromosome Evolution. eLife 2016;5:e17365 doi: http://dx.doi.org/10.7554/eLife.17365 PMID: 27572259 PMCID: 5047749
Meyer, B.J. (2018). Sex and Death: From Cell Fate Specification to Dynamic Control of X-Chromosome Structure and Gene Expression. (E. B. Wilson Award Essay). Mol. Biol. Cell. DOI: 10.1091/mbc.E18-06-0397. PMID: 30376434
Crane, E., Bian, Q., McCord, R.P, Lajoie, B.R., Wheeler, B.S., Ralston, E.J., Uzawa, S., Dekker, J., Meyer, B.J. (2015). Condensin-driven Remodelling of X Chromosome Topology during Dosage Compensation. Nature 523: 240-244. (Epub ahead of print on 1 June 2015) doi: 10.1038/nature14450. PMID: 26030525 PMCID: PMC4498965
Farboud, B. and Meyer, B.J. (2015). Enhancing Genome Editing by CRISPR/Cas9 by Improved Guide RNA Design. Genetics. 199(4): 959-71. doi: 10.1534/genetics.115.175166. [Early online Feb 18, 2015) PMID: 25695951 PMCID: PMC4391549
Severson, A.F. and Meyer, B.J. (2014). Divergent Kleisin Subunits of Cohesin Specify Mechanisms to Tether and Release Meiotic Chromosomes. eLife doi: 10.7554/eLife.03467. PMID: 25171895 PMCID: PMC4174578
Farboud, B., Nix, P., Jow, M.M., Gladden, J.M., Meyer, B.J. (2013). Molecular Antagonism between X-Chromosome and Autosome Signals Determines Nematode Sex. Genes Dev. 27: 1159-1178. PMID: 2366922 PMCID: PMC3672649
Mets, D.G. and Meyer, B.J. (2009). Condensins Regulate DNA Break Distribution and thus Crossover Frequency by Controlling Chromosome Structure. Cell 139: 73-86. PMID: 19781752 PMCID: PMC2785808
Last Updated 2019-01-23