Randy Schekman

Howard Hughes Medical Institute Investigator, Professor of Cell Biology, Development and Physiology*
*And Affiliate, Division of Biochemistry and Molecular Biology
Research Interests
Our research is devoted to a molecular description of the process of membrane assembly and vesicular traffic in eukaryotic cells. Basic principles and approaches that we developed from studies on a simple eukaryote, Saccharomyces cerevisiae, are now being applied to investigate the mechanisms of intracellular vesicular traffic and the biogenesis and function of extracellular vesicles (exosomes) in cultured human cells.
Current Projects
Vesicle Transport Early in the Secretory Pathway
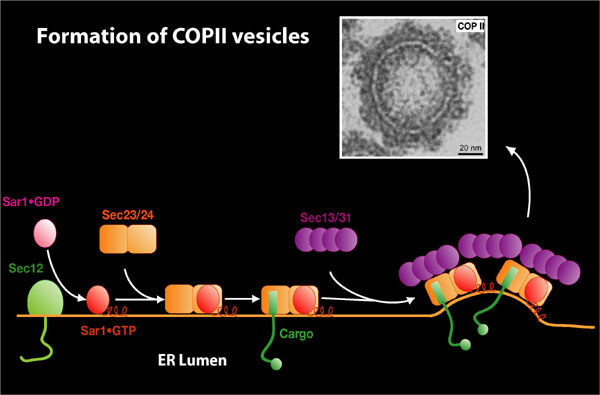
We have developed biochemical assays that measure the early events of polypeptide translocation into the endoplasmic reticulum (ER) and of vesicle-mediated protein transport from the ER to the Golgi apparatus. Vesicles formed in the transport reaction have an electron-dense, 10-nm coat structure that consists of the Sec proteins (the GTP binding protein Sar1p, Sec23/24p, and Sec13/31p) required in budding. This coat (COPII) resembles another coat complex (COPI) that creates transport vesicles within the Golgi apparatus. Our working model is that the Sec protein subunits of the COPII coat bind to the ER membrane and recruit cargo molecules into a cluster that then dimples the membrane to form a bud. A direct interaction between one of the COPII subunits, Sec24p, and membrane proteins is implicated in the capture of cargo proteins. This capture results in the concentrative sorting of membrane and secretory proteins, the latter being selected by an indirect interaction mediated by various membrane receptor proteins that link the coat to soluble cargo proteins. Fission of the bud from the membrane separates transported from resident proteins (1).
Traffic of large and unusually shaped cargo complexes such as procollagen (PC1, 300 nm rod- shaped oligomeric protein) and lipoprotein particles (carrying apolipoprotein B and cholesterol) display special requirements to form larger-than-normal COPII coat complexes. Work in collaboration with the Rape lab led to the discovery of a role for ubiquitylation of the Sec31 subunit of the COPII coat in the capture of procollagen into large COPII vesicles (2). These large, PC1- containing vesicles have been observed in cultured human cells, and in a cell-free COPII vesicle budding reaction (3). The capture of PC1 into larger-than-normal COPII vesicles may be directly influenced by the co-packaging of the Sar1 guanine exchange factor (GEF), Sec12, bound to the ER membrane receptor for PC1, TANGO1 (4). In addition, lipoprotein sorting from the ER appears to require the intervention of a fatty acid binding protein, FABP5, as well as the COPII machinery for particle packaging into transport vesicles (5).
We have also reported a connection between the COPII coat and the formation of the autophagosome, an organelle responsible for the capture of protein aggregates and organelles that are delivered to and degraded in the lysosome. By following the covalent attachment of a lipid group, phosphatidylethanolamine, to the C-terminus of a cytoplasmic protein, LC3, we found that an organelle that mediates vesicular flow between the ER and the Golgi apparatus, the ERGIC, is the membrane that gives rise to an autophagosome precursor organelle (6). Further, we found that the ERGIC forms novel COPII vesicles that build the autophagosome membrane (7).
Unconventional Secretion
In recent years, my research program has focused on the mechanism of unconventional secretion of proteins of physiologic interest such as IL-1 beta (8) and FABP4 (9). Most recently, we have become interested in the spread of α-synuclein (α-syn).
Current effort on α-syn, a protein genetically implicated in Parkinson’s Disease (PD), is focused on how this soluble, relatively unstructured protein may be translocated across a membrane without a typical signal peptide. Alpha synuclein is a major constituent of Lewy Body particles that appear in the dopaminergic neurons in patients who have died of PD. One question is how synuclein may progress out of dopaminergic neurons, in a midbrain tissue called the substantia nigra, into other areas of the brain as may happen in the spread of dementia in some patients with PD. Three different avenues of such intercellular movement are possible: secretion of soluble monomer or oligomer and endocytic reuptake by neighboring cells followed by cytoplasmic penetration to engage the intracellular pool of α- synuclein and another cycle of unconventional secretion; capture of α- synuclein into exosomes that form by budding of vesicles into an endosome followed by secretion of vesicles, endocytic reuptake and cytoplasmic penetration; transmission through intercellular tubular connections, sometimes called tunneling nanotubes, that permit cytoplasmic forms of synuclein monomer or oligomer to spread independent of a membrane penetration event.
In studies using a standard human cell line, HEK293, and with differentiated mouse and human dopaminergic neurons, we found that α-syn is secreted into the growth medium as a soluble monomeric protein, not enclosed in extracellular vesicles. The membrane translocation event appears to involve capture of α-syn into the interior of an endosome in a reaction that requires a palmitoylated form of a membrane anchored DNAJC5 protein (10). The nature of this membrane translocation event remains unknown. It may occur directly across the endosomal membrane or may require the internalization of a complex of α -syn bound to membrane tethered DNAJC5 segregated into exosomes within the endosome. We find that α-synuclein fused to a protein designed to resist unfolding remains competent for secretion, thus a translocation channel would have to be unusually large and flexible, or the process must involve some possibly membrane rupturing event that may occur at the moment of internalization of a vesicle budding into the endosome.

Exosomes and Intercellular Tubular Connections
An entirely distinct form of unconventional secretion invokes the capture of cytoplasmic proteins and small RNA molecules into vesicles exported into the extracellular medium in cultured cells or the extracellular fluids in metazoan organisms. Extracellular vesicles (EVs) form in two distinct pathways. Microvesicles bud directly from the cell surface into the extracellular medium whereas a distinct subset of EVs are formed by membrane invagination into the interior of a late endosome to create a multivesicular body (MVB) which by fusion at the cell surface releases a bolus of EVs called exosomes. Much interest has developed around the evidence that EVs may serve as a means of intercellular communication in which proteins and small RNAs are delivered by fusion or uptake into target cells to mediate control of metabolism or gene expression. Of particular interest is the evidence that the microRNA content of EVs in the blood may change as a result of metastatic cancer and that EVs produced abundantly by tumor cells may communicate with other tissues in the body to create a premetastatic niche into which primary tumor cells may migrate to form a secondary metastatic event (11).

We have devised fractionation procedures to isolate distinct populations of EVs from normal and tumor cells grown in culture. Two vesicle types produced by a human breast cancer cell line are resolved on a buoyant density gradient and each contains a largely non-overlapping set of miRNAs (12). One vesicle type contains several highly selected mature miRNAs which we estimate are ~1000X enriched over their content in the cytoplasm of the tumor cell. A human embryonic cell line produces exosomes that also have several highly selected miRNAs but these differ from those enclosed within the selective pool of vesicles produced by the breast cancer cells (13, 14). These results suggest a cell type-specific high-fidelity sorting reaction to secrete only a subset of miRNAs from cells. This sorting may serve the purpose of selective disposal of certain miRNAs or the targeted delivery of selected miRNAs for control of gene expression in cells that take up and functionally incorporate the exosome content.
Little is known about the regulation of MVB fusion at the cell surface vs the lysosome. Recently we reported the surprising finding that MVB fusion at the cell surface is dramatically stimulated by conditions that damage the plasma membrane. Plasma membrane repair processes have been known for over a century and a repair process has been shown to involve the mobilization of mature lysosomes which fuse at point of rupture to patch damage at the cell surface (15). We reported that the same conditions and indeed some of the same Ca2+-binding proteins are involved in mobilizing MVBs and lysosomes for plasma membrane repair. These conditions greatly stimulate exosome secretion and may offer at least a partial explanation for the abundance of these vesicles in all bodily fluids.
Exosomes and EVs in general have been promoted as a vehicle for intercellular communication. Unfortunately, the evidence supporting this claim is indirect and very weak. For an EV/exosome to be an efficient cargo delivery vehicle, the luminal content of a vesicle must be discharged into the cytoplasm and possibly then into the nucleus of a target cell. This requires a membrane fusion reaction either at the cell surface or after the EV/exosome is internalized into an endosome. Enveloped RNA viruses which have membrane fusion protein catalysts integral to the virus particle serve as the precedent for this kind of delivery. No such membrane fusogen has been demonstrated in any of the published claims of functional cargo delivery by EV/exosomes. Indeed, the efficiency with which cargo is delivered by EVs has not been properly quantified and it remains possible to explain the many effects that have been reported by invoking a very inefficient delivery process.
We have developed two major findings with respect to efficient cargo delivery by EVs. We found that EVs secreted by neuroblastoma or mouse ES cells differentiating into neuronal precursor cells were taken up by and stimulated the rate of neuronal differentiation of mouse ES cells (16). EVs isolated from differentiating neurons were found to fairly selectively sort cyclinD1 and CDK4. Further, cyclinD1 was found to be rate-limiting and essential to speed the rate of differentiation of mouse ES cells. Using a proximity labeling approach, we showed that cyclinD1 in EVs reaches the nucleus in target cells to biotinylate two different nuclear proteins implicated in neuronal differentiation. Although this work did not define a fusogen or accurately quantify the efficiency of cyclinD1 delivery, the results clearly pointed to a mechanism for functional delivery of EV content to a target cell. This may be a specialized case where functional delivery of EV content serves an important role in stem cell differentiation.
We devised a different assay to measure the efficiency of cargo delivery mediated by exosomes engineered to tether Cas9 indirectly to an endosome/exosome-specific membrane protein, CD63 (17). Exosomes isolated from HEK293 cells were 15-fold enriched in enzymatically active Cas9 and a gRNA expressed in the same cells. A recipient cell line was constructed using MDA-MB-231, a breast cancer cell line, expressing a crippled form of Nluc with a stop codon and a single nucleotide deletion introduced at the beginning of the reading frame just after a target sequence for the gRNA. Expression of Cas9 and the gRNA directly in the recipient cell resulted in error-prone repair at the site of ds cleavage sufficient to restore the reading frame and increase Nluc expression 60-100 fold. Exosomes isolated from donor cells were measurably internalized by recipient cells, but very little activation of Nluc gene expression was observed. In contrast, when donor and recipient cells were co-cultured with physical contact, substantial transfer of Cas9 and activation of Nluc expression in recipient cells was observed. Using fluorescence visualization and correlative light and electron microscopy (CLEM), we observed transfer mediated by open-ended membrane tubular connections between HEK2893 and MD-231 cells (fluorescence image below). The formation of such open- ended connections required the action of a membrane fusogen, syncytin, which is expressed in the recipient tumor cell line. Syncytin is normally expressed and functions only at the pre-implantation stage of embryonic development. However, its expression in tumors may suggest a role in tumor growth or metastasis, possibly through intercellular tubular connections or in exosomes secreted by tumor cells.

Selected Publications
-
COPII and the regulation of protein sorting in mammals. Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. Nat Cell Biol. 2011 Dec 22;14(1):20-8. doi: 10.1038/ncb2390. Review. Erratum in: Nat Cell Biol. 2012 Feb;14(2):221.
-
Ubiquitin-dependent regulation of COPII coat size and function. Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgärtel C, Schekman R, Rape M. Nature. 2012 Feb 22;482(7386):495-500. doi: 10.1038/nature10822.
-
COPII-coated membranes function as transport carriers of intracellular procollagen I. Gorur A, Yuan L, Kenny SJ, Baba S, Xu K, Schekman R. J Cell Biol. 2017 Jun 5;216(6):1745-1759. doi: 10.1083/jcb.201702135. Epub 2017 Apr 20.
-
TANGO1 and SEC12 are copackaged with procollagen I to facilitate the generation of large COPII carriers. Yuan L, Kenny SJ, Hemmati J, Xu K, Schekman R.Proc Natl Acad Sci U S A. 2018 Dec 26;115(52):E12255-E12264. doi: 10.1073/pnas.1814810115. Epub 2018 Dec 13.
-
Fatty-acid binding protein 5 modulates the SAR1 GTPase cycle and enhances budding of large COPII cargoes. Melville D, Gorur A, Schekman R. Mol Biol Cell. 2019 Feb 1;30(3):387-399. doi: 10.1091/mbc.E18-09-0548. Epub 2018 Nov 28.
-
The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Ge L, Melville D, Zhang M, Schekman R. Elife. 2013 Aug 6;2:e00947. doi: 10.7554/eLife.00947.
-
Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Ge L, Zhang M, Schekman R. Elife. 2014 Nov 28;3:e04135.
-
Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Elife. 2015 Nov 2;4. pii: e11205.
-
Unconventional secretion of FABP4 by endosomes and secretory lysosomes.
Villeneuve J, Bassaganyas L, Lepreux S, Chiritoiu M, Costet P, Ripoche J, Malhotra V, Schekman R. J Cell Biol. 2018 Feb 5;217(2):649-665. doi: 10.1083/jcb.201705047. Epub 2017 Dec 6.
-
Unconventional secretion of α-synuclein mediated by palmitoylated DNAJC5 oligomers.
Wu S, Hernandez Villegas NC, Sirkis DW, Thomas-Wright I, Wade-Martins R, Schekman R. Elife. 2023 Jan 10;12:e85837. doi: 10.7554/eLife.85837. PMID: 36626307.
-
Extracellular Vesicles and Cancer: Caveat Lector Matthew J. Shurtleff, Morayma M. Temoche-Diaz, Randy Schekman. Annual Review of Cancer Biology 2018 2:1, 395-411.
-
Distinct mechanisms of microRNA sorting into cancer-derived extracellular vesicle subtypes. Temoche-Diaz, M., Shurtleff, M.J., Nottingham, R., Yao, J., Fadadu, R. P., Lambowitz, A.and Schekman, R. (preprint posted on bioRxiv: http://biorxiv.org/cgi/content/short/612069v1).
-
Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Shurtleff MJ, Temoche-Diaz MM, Karfilis KV, Ri S, Schekman R. Elife. 2016 Aug 25;5. pii: e19276. doi: 10.7554/eLife.19276.
-
Broad role for YBX1 in defining the small noncoding RNA composition of exosomes.
Shurtleff MJ, Yao J, Qin Y, Nottingham RM, Temoche-Diaz MM, Schekman R, Lambowitz AM.Proc Natl Acad Sci U S A. 2017 Oct 24;114(43):E8987-E8995. doi: 10.1073/pnas.1712108114. Epub 2017 Oct 10.
-
Annexin A6 mediates calcium-dependent exosome secretion during plasma membrane repair.
Williams JK*, Ngo JM* , Lehman IM, Schekman R. (*equal contribution) Elife. 2023 May 19;12:e86556. doi: 10.7554/eLife.86556. PMID: 37204294.
-
Extracellular vesicles from neurons promote neural induction of stem cells through cyclin D1.
Song L, Tian X, Schekman R. J Cell Biol. 2021 Sep 6;220(9):e202101075. doi: 10.1083/jcb.202101075. PMID: 34309628.
-
Syncytin-mediated open-ended membrane tubular connections facilitate the intercellular transfer of cargos including Cas9 protein.
Zhang C, Schekman R. Elife. 2023 Mar 10;12:e84391. doi: 10.7554/eLife.84391. PMID: 36896791.
Last Updated 2024-06-05