Faculty Research Page

Howard Hughes Medical Institute Investigator and Chancellor's Adjunct Professor of Biochemistry, Biophysics and Structural Biology
Research Interests
Current Projects
The Epidermal Growth Factor Receptor (EGFR): We made a breakthrough in understanding how the epidermal growth factor receptor (EGFR) is activated, leading to major shifts in how the field views kinase activation by dimerization. We showed that activation of EGFR involves the formation of an asymmetric dimer between the kinase domains of the receptor, in which one kinase domain serves as an activator of the other, analogous to the activation of cyclin-dependent kinases by the cell cycle control proteins known as cyclins (Zhang et al., Cell 2006 ; Zhang et al., Nature 2007). This new paradigm for the activation of a receptor tyrosine kinase explained why two human EGFR family members, HER2 and HER3, are particularly potent in combination even though HER3 lacks catalytic activity. The interaction between HER2 and HER3 underlies many cancers, including glioblastomas, and the results from our group provided the first clear mechanistic insights into how these two receptors form an active signaling complex.
We also demonstrated that the cytoplasmic module of EGFR is capable of dimerizing and activating on its own, because the juxtamembrane segment of the receptor is able to dimerize the kinase domains (Jura et al., Cell 2009; this was also shown independently by Mark Lemmon and colleagues, University of Pennsylvania). Data from our group are consistent with a mechanism in which the extracellular domains prevent the intrinsic ability of the transmembrane and cytoplasmic domains to dimerize and activate, with ligand binding releasing this block. Recently, we used cell-based analyses to work out the nature of the coupling between the extracellular domains of the receptor and the kinase domains, demonstrating that the extracellular binding of ligands to the receptor is transmitted to the intracellular kinase domains through a specific conformational coupling of the transmembrane helices (Endres et al., Cell 2013).Towards the goal of defininghow EGFR transduces signals, we determined the structure of the transmembrane and juxtamembrane segments of the receptor in a lipid bilayer. This led to a model for how ligand binding activates the intracellular kinase domains. We studied the stoichiometry of the receptor and showed that activation converts monomeric EGFR not only to dimers, but also to higher-order multimers. We developed a molecular model for these multimers, to understand how they boost the EGF response.
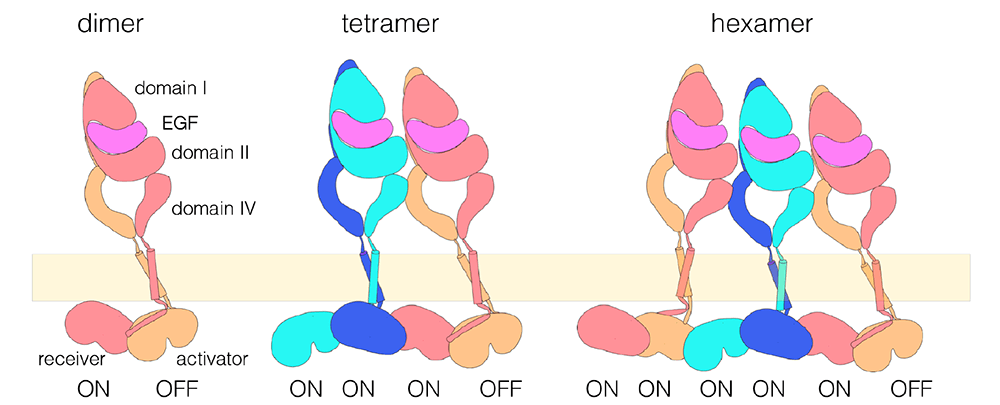
Figure: A model for EGFR multimerization. The schematic model for a dimer is based on crystal structures of the extracellular and intracellular modules, and NMR structures of the transmembrane and juxtamembrane segments (Kovacs et al., 2015). The model for an EGFR tetramer is based on computational docking of extracellular dimers, followed by connection to the structure of the dimeric transmembrane helices and a chain of asymmetric dimers of kinase domains (Huang et al., 2016). Higher-order multimers might be generated by repeating the interactions used to generate the model for the tetramer.
The activation of Ras: Ras is a critical ignition switch in signaling by cell-surface receptors (Vetter and Wittinghofer, 2001), and we seek to understand how it is activated at the membrane. We are following two complementary streams of research to understand how Ras is regulated, and how it malfunctions in disease. We are reconstituting membrane-associated forms of Ras with its regulators and effectors, for functional and structural studies. To gain further insight into mechanism and evolution, we are using high-throughput methods to understand how mutations modulate Ras function.
Recently, we determined the structural mechanisms underlying the interaction of Ras with one class of nucleotide exchange factors, the RasGRP proteins. A new understanding of the sensitivity of Ras to mutations has emerged from studies using deep mutagenesis, which allowed us to map the mutational landscape of Ras comprehensively. Moving forward, we have reconstituted Ras with activators and effectors on membranes for mechanistic studies and cryoEM analysis. Our data on the mutational sensitivity of Ras indicate that the protein gained tighter allosteric control in the vertebrate lineage, and we are tracing the evolutionary history of allosteric control in Ras to understand the origin of this effect.
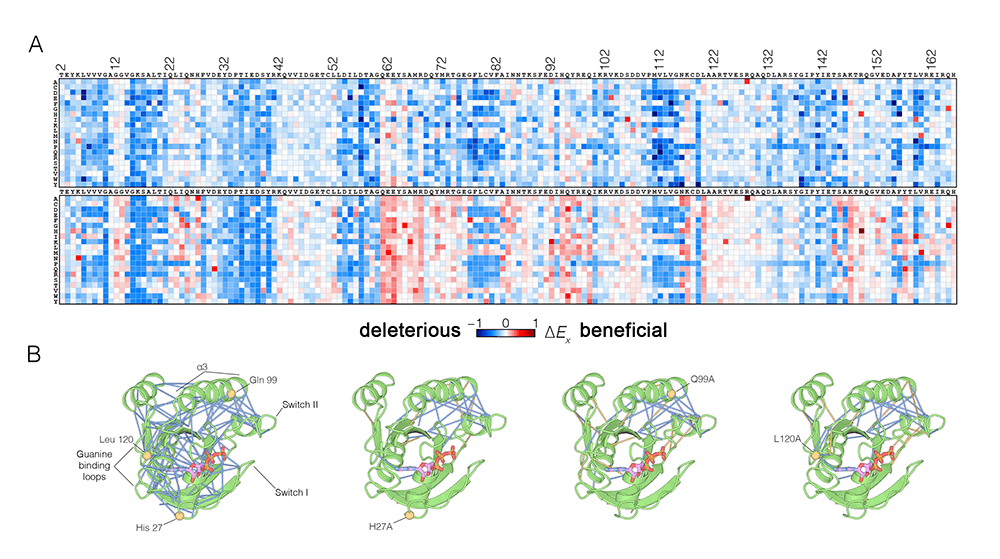
Figure: Effects of mutations on Ras. (A) Mutational tolerance of Ras in the presence (top) and absence (bottom) of the GAP and GEF regulators (Bandaru et al., 2017). The results are shown in the form of a 165 x 20 matrix. Each row of the matrix represents one of the 20 amino acids, and each column shows one of the residues of Ras, from 2 to 166. Each entry in the matrix represents the value of the relative enrichment for the corresponding mutation. In the presence of regulators, most mutations are deleterious. The absence of the regulators is marked by several hotspots of activating mutations (red). (B) The impact of hotspot mutations on structural flexibility in Ras. The panel shows sustained sidechain-sidechain contacts in molecular dynamics simulations (0.6 ms each) of wild-type Ras.GTP and three hotspot mutants. Strong interactions between sidechains are identified by blue lines. For simulations of mutant Ras, the blue lines indicate contacts that are also present in the wild-type simulation. The yellow lines indicate contacts that are unique to the mutant simulation. Note that the network of strong contacts present in the wildtype simulation is disrupted similarly in the three simulations of Ras mutants.
Tyrosine kinase signaling in immune cells: We are studying the regulation of the cytoplasmic tyrosine kinases that transmit signals from T cell and B cell receptors. These include Lck (a Src-family kinase; Lyn in B cells), ZAP-70 (Syk in B cells), and Itk (a Tec-family kinase; Btk in B cells). Here too, we are following a strategy that uses reconstituted systems to monitor function, in correlation with high-throughput mutagenesis to survey the effects of sequence variation. We have developed a method to determine the specificity profiles of tyrosine kinases, based on bacterial surface display of peptides and deep sequencing. Using this, we discovered an unexpected degree of specificity in ZAP-70 and Lck. Our results modify the view that tyrosine kinase specificity arises primarily from localization effects. For the Tec family of tyrosine kinases, we determined the mechanism by which the lipid-binding PH-TH module regulates the activity of Btk, an important drug target in cancer.
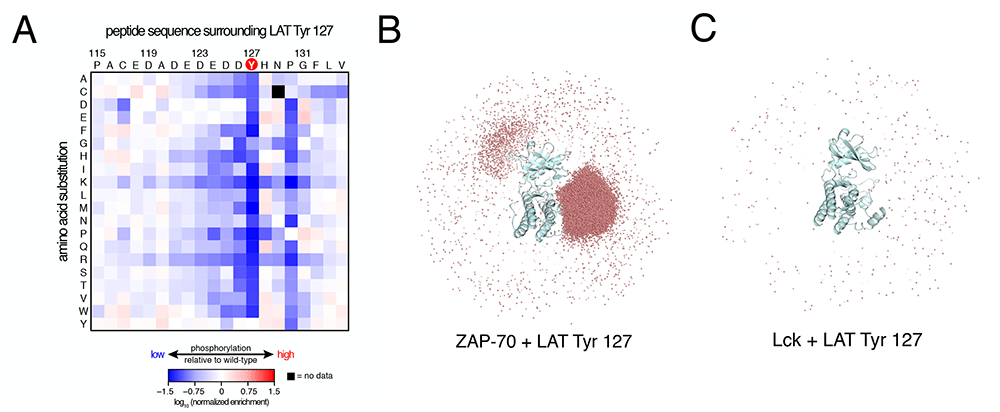
Figure: An electrostatic filter in ZAP-70 (Shah et al., 2016). (A) Saturation mutagenesis of a LAT peptide with screening for phosphorylation by ZAP-70, with blue corresponding to low phosphorylation. Note that the occurrence of arginine or lysine at almost any position attenuates phosphorylation. (B) Illustration of electrostatic effects in substrate selection from Brownian dynamics simulations of LAT peptide association with ZAP-70. The red dots represent the positions of the peptide during the simulations, with dense clustering near the active site. (C) As in panel B, for the same peptide associating with Lck.
Activation-triggered subunit exchange in CaMKII: CaMKII is a calcium-dependent kinase that plays crucial roles in neuronal and cardiac signaling, by phosphorylating ion channels. CaMKIIhas a unique architecture, in which the catalytic domains are linked flexibly to a hub domain, which forms a dodecameric or tetradecameric ring (Chao et al., 2011). This organization enables the highly cooperative activation of CaMKII by Ca2+/calmodulin (Ca2+/CaM). We discovered recently that CaMKII holoenzymes undergo activation-triggered subunit exchange, such that subunits from activated holoenzymes can transphosphorylate and activate subunits from unactivated holoenzymes. We are currently developing cell-based fluorescence imaging assays to monitor subunit exchange, in order to study the physiological relevance of this phenomenon.
DNA Polymerase Clamp Loaders: Highly processive DNA polymerases are tethered to ring-shaped sliding clamps that encircle DNA, allowing rapid movement of the replication fork (Kelch et al., 2012). Over two decades, I worked with Mike O’Donnell (Rockefeller University) to establish the structural principles underlying the clamp-loader complexes that load the sliding clamps onto DNA. These machines have ATPase subunits that respond allosterically to ATP, to each other, and to DNA. These subunits are related evolutionarily to Ras, providing a conceptual connection to our projects on signaling.
Previously, we had determined structures of clamp-loader complexes in various states of assembly, but not in complete form. We put this all together by determining structures of complete clamp-loader/clamp complexes on DNA. In a new direction for this project, we plan to use deep mutagenesis and protein engineering to gain a definitive understanding of how ATP hydrolysis is coupled to DNA recognition, with the ultimate goal of relating clamp-loader mechanism to that of DNA translocases.
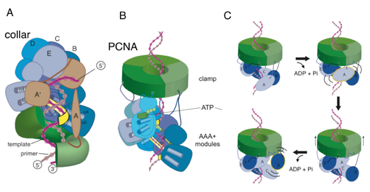
Figure: Structure of the clamp-loader complex and speculative design of a DNA translocase based on clamp loader subunits. (A) Crystal structure of the T4 clamp loader, bound to a primer-template junction and an open clamp (Kelch et al., 2011). The clamp loader consists of 4 active AAA+ ATPases (blue) and one that has degenerated in structure (brown). The AAA+ modules spiral around the DNA, as does the clamp, which is open. The AAA+ modules only make contact with the template strand (yellow). The interfacial ATP binding sites are primed for catalysis, but the structure is trapped by the use of ADP.BeFx. (B) The starting point for experimentation aimed at building a translocase from clamp-loader subunits. In this hypothetical example, six copies of clamp loader AAA+ modules are fused to the trimeric PCNA sliding clamp, using a strategy used to produce SUMO-modified PCNA (Freudenthal et al., 2011). The collar of the clamp loader is replaced by PCNA, so that DNA bound by the ATPase subunits (shades of blue) can thread through PCNA. (C) Six identical AAA+ modules attached to a sliding clamp on DNA will form a filament in the presence of ATP. Hydrolysis at the first AAA+, labeled A, causes it to detach from DNA. The last AAA+ in the filament, F, cannot hydrolyze ATP since it is not at an interface. It can capture the newly released A subunit, thus moving the sliding clamp up by ~2.5 base pairs. This process can repeat for directional movement.
Selected Publications
For a complete list of Kuriyan lab publications, visit:
https://www.ncbi.nlm.nih.gov/myncbi/browse/collection/41158434/?sort=date&direction=descending
For descriptions of research articles from the lab visit:
https://jkweb.berkeley.edu/publications/
Last Updated 2019-02-06