Jasper Rine

Professor Emeritus of Genetics, Genomics, Evolution, and Development
Research Interests
The research in my lab is centered on the epigenetic mechanisms by which the information in a genome is expressed in a manner that is stable and heritable through cell division. This issue has led us to discoveries on how specialized domains of chromatin structure are established, maintained, and inherited. Our studies use the yeast Saccharomyces cerevisiae and the powerful genetic, genomic, and proteomic approaches that are so facile in this organism to study the nature of the chromatin in specialized domains of the yeast genome, how it is assembled and modified, and how these modifications affect its expression, replication, and repair. Recently, the lab has started a new emphasis focused on exploring the functional consequences of human genetic and epigenetic variation with the goal of understanding and reducing the incidence of two of the most common birth defects, and the mechanism of particular driver mutations in human cancer.
Current Projects
The most prominent epigenetic mechanism in Saccharomyces involves the silencing of genes controlling cell type. The a and alpha cell types of yeast are controlled by transposable genes that are alleles of the mating type locus (MAT). These alleles are complex blocks of DNA encoding regulatory proteins whose synthesis controls the expression of genes that specify the identity of different cell types of yeast. In addition to MAT, the a and alpha genes are also located at two additional loci, known as HML and HMR, where they are transcriptionally silenced. Transcriptional silencing requires a complex series of interactions between proteins, regulatory sites, and origins of replication that result in the establishment of inactive chromatin in these regions. This repression mechanism is one of the best examples in biology to learn how changes in chromatin structure cause changes in gene expression.
We have focused our efforts on the Sir proteins and related sirtuins, required for this position-dependent regulation, and on the silencers, which are sites required for the silencing of flanking sequences. Repression mediated by the Sir proteins involves the assembly of inactive chromatin (heterochromatin) at HML and HMR. This repressive chromatin structure, once established, is epigenetically inherited, and offers the most tractable opportunity to study the inheritance of states of chromatin structure. Remarkably, the silencer also functions as a chromosomal origin of DNA replication, and we have discovered that ORC, the eukaryotic replication initiator, PCNA and other replication-related proteins play critical roles in silencing.
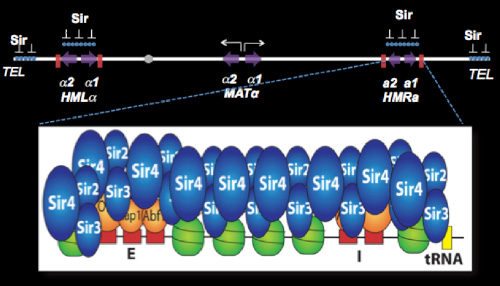
Silencing the HML and HMR loci requires passage of cells through the cell cycle. We have shown that Sir proteins can bind to silencers and spread through these regions, binding to nucleosome tails, in cells that are in G1, but silencing is not achieved until later. By studying silencing kinetically with single-cell resolution, we have discovered that multiple cell cycles are required to fully silence these loci, even though the structural hallmarks of heterochromatin, are laid down in the first cell cycle. Our goal is a complete description of the cell-cycle-dependent steps involved in making silenced chromatin.
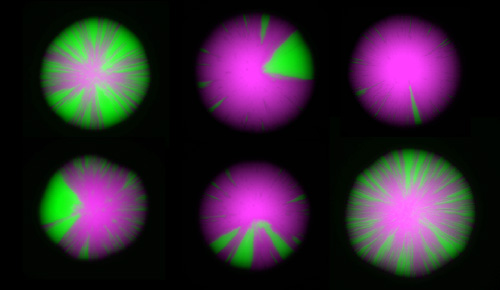
The frontiers of chromatin research include measurements of the dynamics of chromatin, and studies at the single-cell level. We have recently devised an elegant assay revealing dynamics at the single-cell level that allows us to detect rare and even transient perturbations of chromatin structures by linking them to the creation of a permanent and heritable mark: a switch from the expression of RFP to the expression of GFP. As a colony of cells grow, chromatin dynamics in individual cells are revealed by the appearance of green sectors on otherwise red colonies. This assay has led to a flurry of new discoveries, enabling us to assemble a nearly complete description of all the factors that influence the dynamics of gene repression. This image highlights the impact of mutations in various chromatin-modifying enzymes on chromatin dynamics.
The interface between metabolism and chromatin structure is emerging as an exciting area in both basic research and in cancer biology. One example is the discovery of isocitrate dehydrogenase mutations as driver mutations in many different human cancers. One hypothesis for how these mutations promoter cancer is through production of an onco-metabolite, 2-hydroxyglutarate (2HG), with the potential to inhibit a-ketoglutarate- dependent dioxygensases, such as the human TET enzymes, which demethylate DNA. We have shown that the very same isocitrate dehydrogenase mutations, when introduced into yeast, cause the accumulation of 2-HG, and result in epigenetic consequences. Importantly, yeast lack DNA methylation. Therefore there must be a different mechanism(s) for this epigenetic impact, which we are now zeroing in on.
Amazingly, a single amino acid change in a particular repressor can restore heterochromatin in cells lacking the usual heterochromatin components. This discovery implies that heterochromatin may be easy to evolve, and indeed the mechanisms of heterochromatin silencing are very different between Saccharomyces and the distantly related Schizzosaccharomyces pombe. We have embarked on a computational and functional exploration of how heterochromatin evolved by exploiting the fantastic insights that can come from the dozens of sequenced genomes from closely and less-closely related yeast species. These studies are already revealing crucial steps in the evolution of this type of chromatin structure and have revealed how silencing has shaped the genetic organization of the genome.
The newest project in the lab is focused on human genetic variation. The defining genetic challenge of this century will be understanding the significance of the 3 to 6 million DNA sequence differences between any two people. Many of these differences will be neutral polymorphisms, others will be biologically important and medically significant. There are hundreds of human proteins that can functionally replace their human orthologs when human cDNAs are expressed in a yeast strain in which the corresponding yeast gene has been removed. In this way, yeast can be used to systematically investigate variation in the protein coding sequences of human genes, free of the complications presented by studying the different mixtures of variants found in any two humans. Our earliest studies have focused on enzymes of folate metabolism due to the central role that folate plays in reducing the incidence of neural tube defects, one of the most common forms of birth defects. To our surprise, we found that approximately half of the nonsynonymous changes found in one of these enzymes from a random sample of humans dramatically affect the function of this enzyme. Remarkably, in most of these cases, the defect in the enzyme's function can be largely suppressed by increasing the concentration of the enzyme's cofactor. If these observations apply broadly, it should be possible to identify those individuals most at risk, and at least in some cases, it may be possible to reduce their risk with appropriate cofactor supplementation.
Nine percent of humans are homozygous for a mutation in folate biosynthesis that reduces the synthesis of the methyl-donor for DNA methylation. We hypothesize that homozygotes for this mutation may have a high frequency of epialleles at those positions of the human genome that are most sensitive to a reduction in the level of methyl donor for DNA methylation. We have shown that limitations in folate metabolism have epigenetic consequences in various yeast species and in human cells in culture. We hypothesize that people with alterations in folate metabolism may be particularly prone to changes in their epigenetic program. Our goal in this project is to discover the genetic basis of folate-preventable birth defects such as spina bifida and cleft palate.
Selected Publications
Dodson AE, Rine J. 2015 Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae. Elife. 2015 Jan 12;4:e05007. PMID: 25581000
Steakley DL and Rine J 2015 On the mechanism of Transcriptional Silencing. Genes, Genomes and Genetics (G3) Jun 16. pii: g3.115.018515. doi: 10.1534/g3.115.018515. PMID: 26082137
Ellahi A, Thurtle, DM and Rine J 2015 The chromatin and transcriptional landscape of native Saccharomyces cerevisiae telomeres and subtelomeric domains. GENETICS 200:505-521 PMID: 25823445
Lombardi LM, Davis MD, Rine J. 2015 Maintenance of nucleosomal balance in cis by conserved AAA-ATPase Yta7. GENETICS. 2015 199:105-16. PMID: 25406467
Thurtle D and Rine J 2014 The molecular topology of silenced chromatin in Saccharomyces. Genes and Development, 28:245-58. PMID: 24493645
Sadhu M, Guan Q, Li F, Sales-Lee J, Iavarone AT, Hammond MC, Cande WZ and Rine J 2013, Nutritional control of epigenetic processes in yeast and human cells. GENETICS 195:831-844 PMID 23979574
Teytelman L, Thurtle D. Rine J and van Oudenaarden A 2013 Highly expressed loci show misleading ChIP localization for multiple unrelated proteins Proc.Natl.Acad.Sci 110:18602-7. PMID: 24173036
Zunder RM and Rine J 2012 Direct interplay among histones, histone chaperones, and a chromatin boundary protein in the control of histone gene expression Molec. Cell. Biol. 32:4337-49. PMID: 22907759
Mayfield, J.A., M.W. Davies, D. Dimster-Denk, E. Boydston, L. Fink, X.X. Lin, S. McCarthy, A. Narian, N. Pleksac and J. Rine. 2012 Surrogate genetics and metabolic profiling for characterization of human disease alleles GENETICS. 190:1309-23 PMID: 22267502.
Zunder RM, Antczak AJ, Berger JM, Rine J. 2012 Two surfaces on the histone chaperone Rtt106 mediate histone binding, replication, and silencing. Proc Natl Acad Sci U S A. 109:E144-53. PMID: 22198837
Lombardi LM, Ellahi A, Rine J. 2011 Direct regulation of nucleosome density by the conserved AAA-ATPase Yta7 Proc Natl Acad Sci 108:E1302-11. PMID: 22074782
Osborne EA, Dudoit S, and Rine J. 2009 The establishment of gene silencing at single cell resolution. Nature Genetics. 41: 800-806. PMID: 19543267
Zill OA and Rine J. 2008 Interspecies variation reveals a conserved repressor of alpha-specific genes in Saccharomyces yeasts. Genes Dev. 22: 1704-16 PMID: 18559484.
Photo credit: Mark Joseph Hanson of Mark Joseph Studio.
Last Updated 2009-06-11