Molecular Networks Controlling Dynamic Chromosome Behaviors during Development
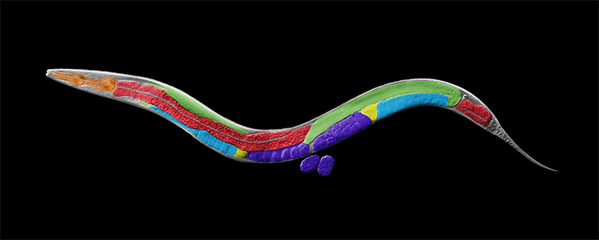
Our research focuses on the interplay between the structure of chromosomes and their function. Chromosomes undergo dynamic behaviors during development to ensure genome stability and accurate cell fate decisions. We study inter-related molecular networks that control diverse chromosome behaviors: chromosome counting to determine sexual fate; X-chromosome remodeling to achieve X-chromosome repression during dosage compensation, an epigenetic process; chromosome cohesion to tether and release replicated chromosomes for reducing genome copy number during germ cell formation; and chromosome compaction to control gene expression, chromosome segregation, and recombination between maternal and paternal chromosomes. We have found that the developmental control of gene expression is achieved through chromatin modifications that affect chromosome structure with epigenetic consequences. We have also established robust procedures for targeted genome editing across nematode species diverged by 300 MYR to study the evolution of sex determination and dosage compensation. We combine genetic, genomic, proteomic, biochemical, and cell biological approaches to study these questions in the model organism Caenorhabditis elegans, a round worm, and its related nematode species.
Counting Chromosomes to Determine Sex: Molecular Antagonism between X-Chromosome, Autosome Signals Specifies Nematode Sex
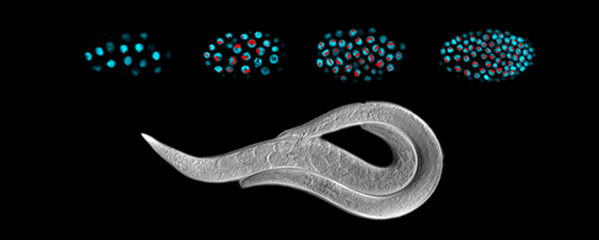
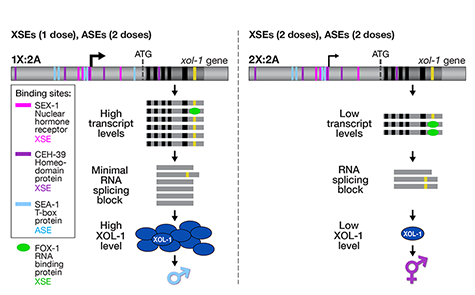
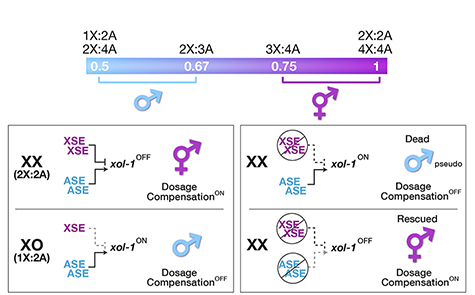
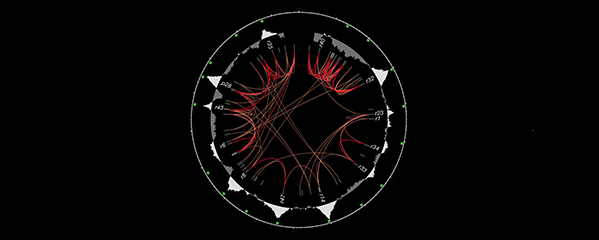
Many organisms determine sexual fate by a chromosome-counting mechanism that distinguishes one X chromosome from two. Embryos with one X become males, while those with two become females. We dissected the molecular mechanism by which the nematode C. elegans counts its sex chromosomes to discern how small changes in the concentrations of molecular signals are translated into dramatically different developmental fates. C. elegans tallies X-chromosome number relative to the ploidy, the
sets of autosomes (X:A signal). It discriminates with high fidelity between tiny differences in the signal: 2X:3A embryos (ratio 0.67) become males, while 3X:4A embryos (ratio 0.75) become hermaphrodites (Figure 1).
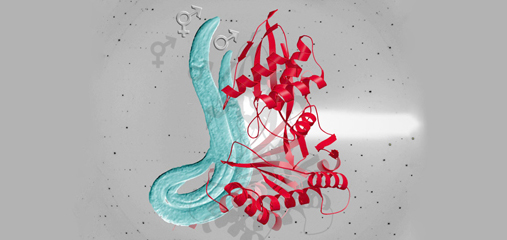
We showed that a set of X-linked genes called X-signal elements (XSEs) communicates X-chromosome dose by repressing the master sex-determination switch gene xol-1 in a cumulative, dose-dependent manner. XOL-1, a GHMP kinase, is activated in 1X:2A embryos (1 dose of XSEs) to set the male fate but repressed in 2X:2A embryos (2 doses of XSEs) to promote the hermaphrodite fate, including the activation of X-chromosome dosage compensation. We also showed that the dose of autosomes is communicated by a set of autosomal signal elements (ASEs) that also act in a cumulative, dose-dependent manner to counter XSEs by stimulating xol-1 transcription. We have explored the biochemical basis by which XSEs counter ASEs to determine sex. Analysis in vitro showed that XSEs (nuclear receptors and homeodomain proteins) and ASEs (T-box and zinc-finger proteins) bind directly to at least 5 distinct sites in xol-1 regulatory DNA to counteract each other's activities and thereby regulate xol-1 transcription (Figure 2). Analysis in vivo showed that disrupting ASE and XSE binding sites recapitulated the mis-regulation of xol-1 transcription caused by disrupting the cognate signal element genes. XSE and ASE binding sites are distinct and non-overlapping, suggesting that direct competition for xol-1 binding is not the mechanism by which XSEs counter ASEs. Instead, XSEs likely antagonize ASEs by recruiting cofactors with reciprocal activities that induce opposite transcriptional states. The X:A balance is thus communicated in part through multiple antagonistic molecular interactions carried out on a single promoter, revealing how small differences in X:A values can elicit different sexual fates. We are currently identifying potential coactivators and corepressors and directing efforts toward understanding the evolution of the X:A signal across nematode species.
Although most XSEs repress xol-1 by regulating transcription, one XSE, an RNA binding protein, represses xol-1 by binding to an alternatively spliced intron and blocking its proper splicing, thereby generating a non-functional transcript with an in-frame stop codon (Figure 2). This second tier of repression enhances the fidelity of the counting process.
The concept of a sex signal comprising competing XSEs and ASEs arose as a theory for fruit flies one century ago, and it subsequently became entrenched in textbooks. Ironically, the recent work of others showed the fly sex signal does not fit this simple paradigm, but our work shows the worm signal does.
X-Chromosome Dosage Compensation: Repressing X Chromosomes via Molecular Machines.
Organisms that use sex chromosomes to determine sexual fate evolved the essential, chromosome-wide regulatory process called dosage compensation to balance X-chromosome gene expression between the sexes. Strategies for dosage compensation differ from worms to mammals, but invariably a regulatory complex is targeted to X chromosomes of one sex to modulate transcription along the entire chromosome. The heritable, regulation of X-chromosome expression during dosage compensation is exemplary for dissecting the coordinate regulation of gene expression over large chromosomal territories and the role of chromosome structure in regulating gene expression.
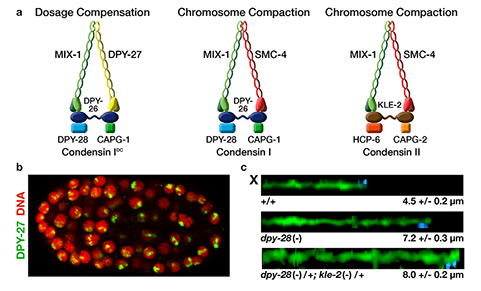
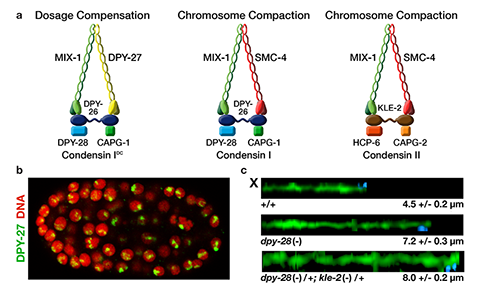
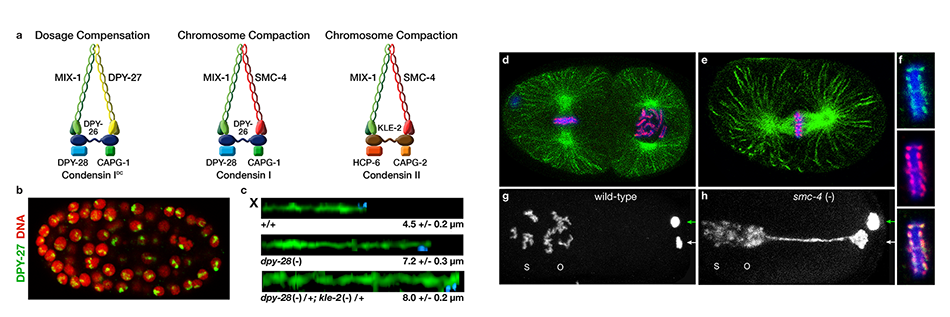
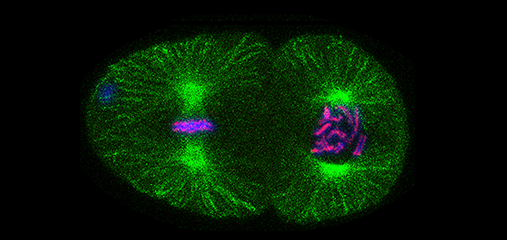
We defined the C. elegans dosage compensation complex (DCC) and showed it is homologous to condensin, a conserved protein complex that mediates the compaction, resolution, and segregation of mitotic and meiotic chromosomes from yeast to humans (Figure 3). The DCC binds to both X chromosomes of hermaphrodites to reduce transcription by half (Figure 3). Failure to reduce expression kills hermaphrodites. Most DCC condensin subunits also control the structure and function of mitotic and meiotic chromosomes by participating in two other distinct condensin complexes (Figure 3). Not only has the DCC co-opt subunits of condensin to control gene expression, it co-opted a subunit from the MLL/COMPASS complex, a histone modifying complex, to help recruit condensin subunits to rex sites.
We found that the DCC condensin subunits are recruited specifically to hermaphrodite X chromosomes by sex-specific DCC subunits that trigger binding to cis-acting regulatory elements on X, called rex and dox sites. rex (recruitment elements on X)
sites recruit the DCC in an autonomous, sequence-dependent manner using DNA motifs highly enriched on X chromosomes. The DCC spreads to dox (dependent on X) sites, which reside in promoters of active genes and bind the DCC robustly only when linked to rex sites.
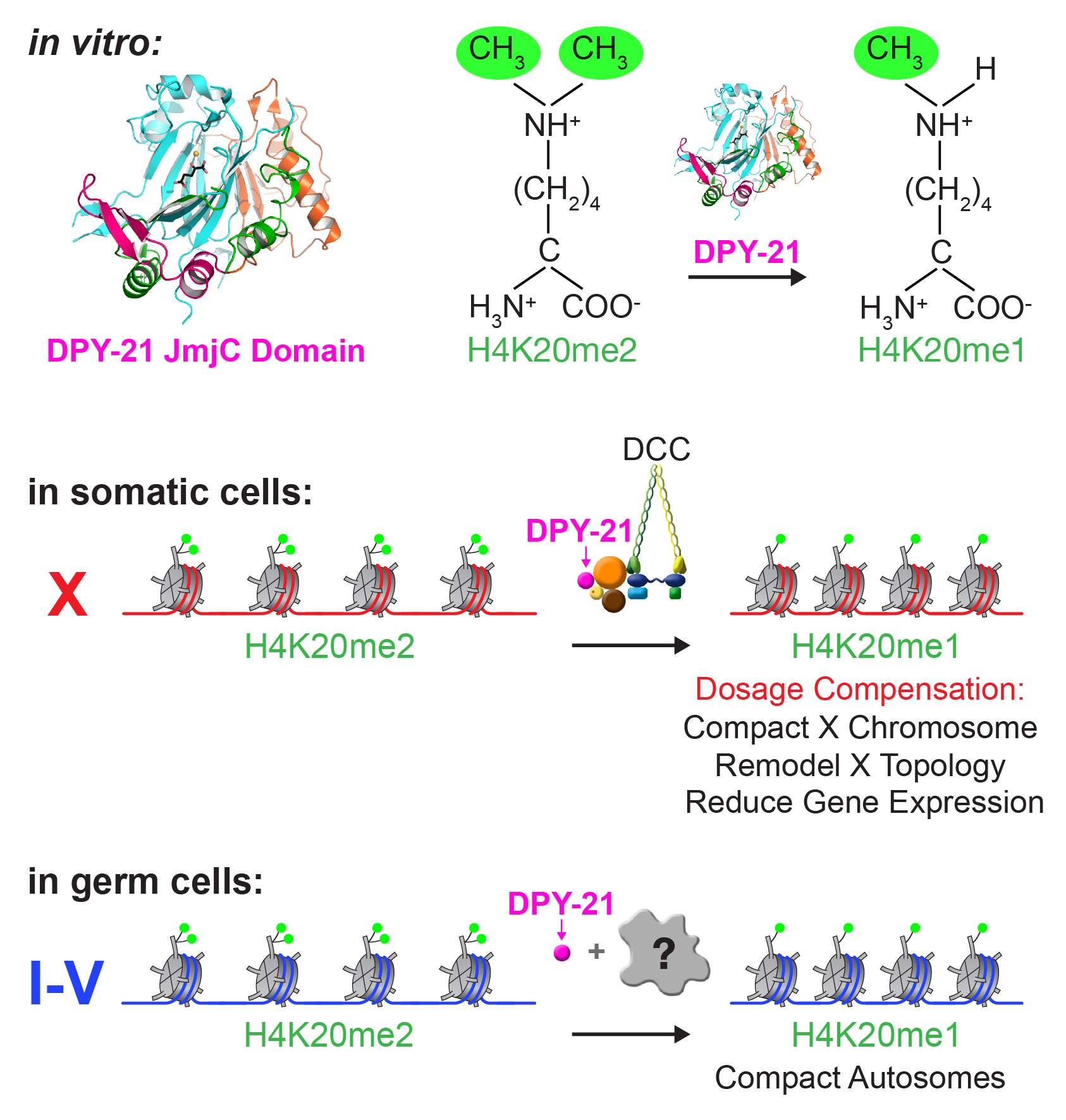
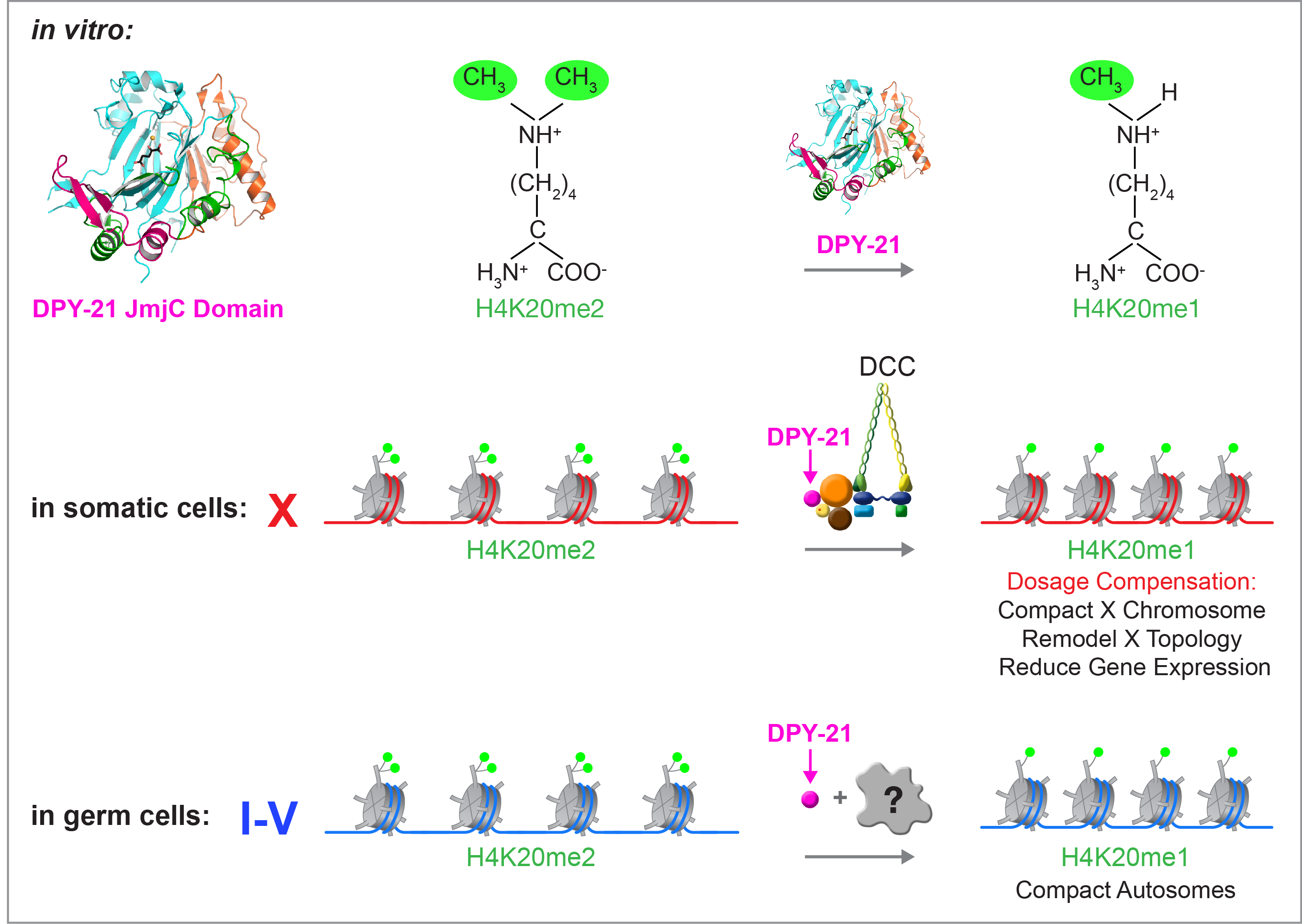
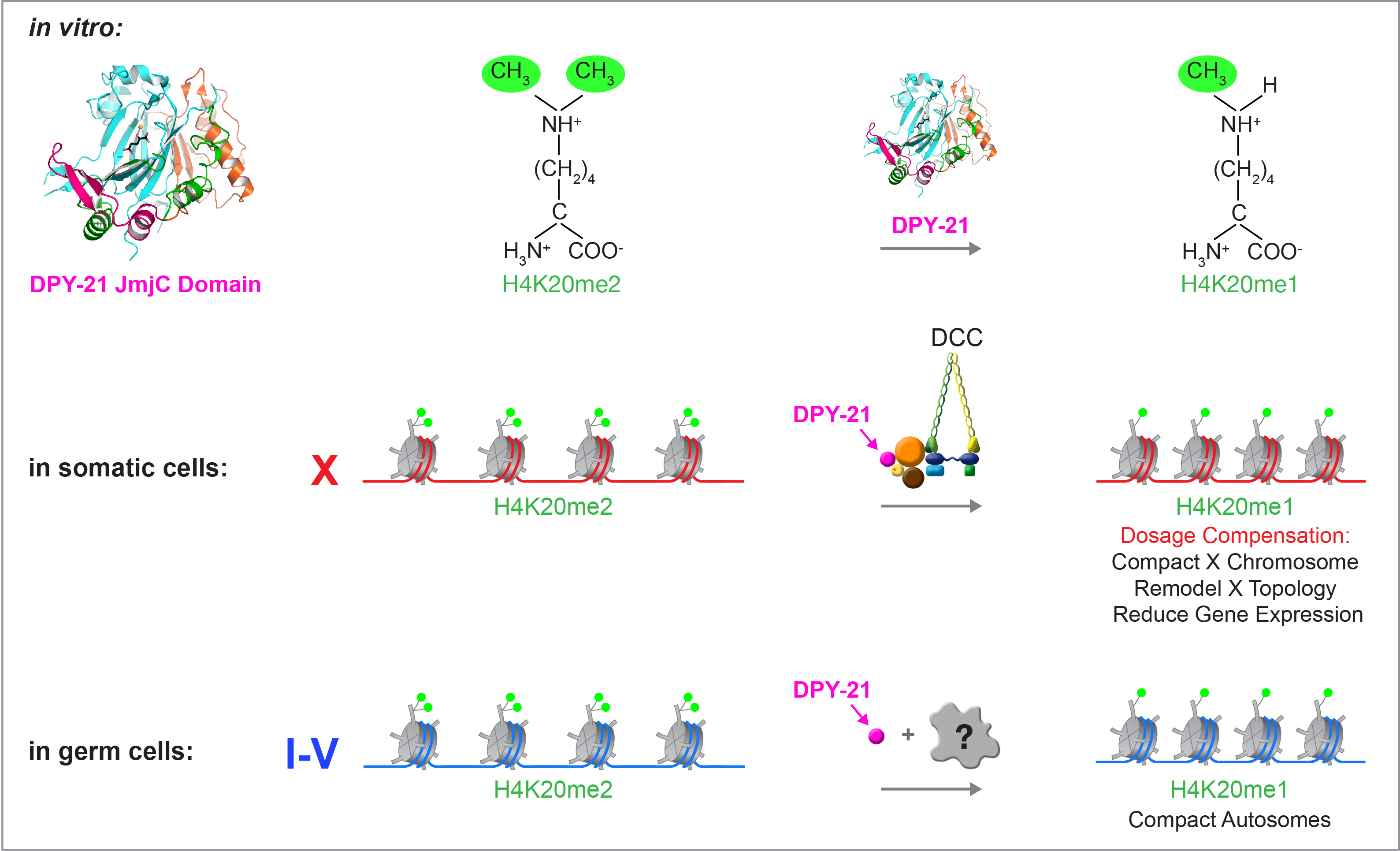
Dynamic Control of X-Chromosome Conformation and Repression by a Histone H4K20me Demethylase.
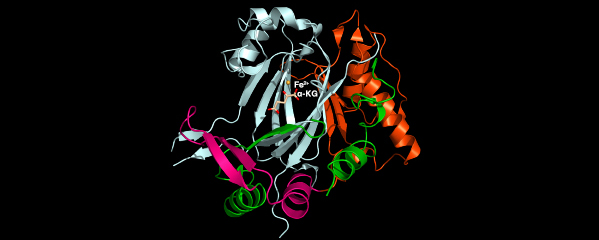
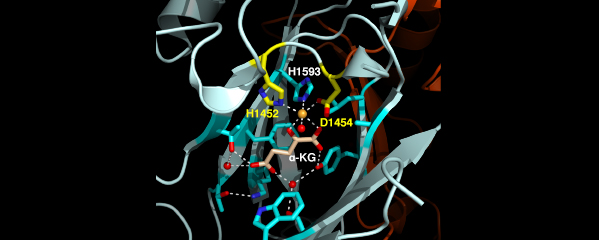
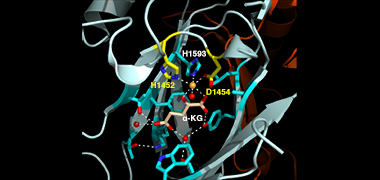
We recently found that DCC subunit DPY-21 has a histone demethylase activity that is responsible for the selective enrichment of H4K20me1 on X chromosomes of XX embryos upon DCC binding. X-ray crystallography and biochemical assays of DPY-21 revealed a novel subfamily of Jumonji C histone demethylases that converts H4K20me2 to H4K20me1. Selective inactivation of demethylase activity in vivo by genome editing eliminated H4K20me1 enrichment on X, elevated X-linked gene expression, reduced X-chromosome compaction, and disrupted X-chromosome topology by weakening TAD boundaries. These findings, among others, demonstrate the direct impact of chromatin modification on higher-order chromosome structure in long-range regulation of gene expression.
H4K20me1 is also enriched on the mammalian inactive X chromosome, but the role of this enrichment in mammalian transcriptional silencing is not known, nor is a selective reagent available to test its role. We showed that the mouse homolog of the DCC subunit also has H4K20me2 demethylase activity. Hence the worm system holds great promise for understanding the effects of histone modification in mammals.
Unexpectedly, DPY-21 associates with autosomes but not X chromosomes of germ cells in a DCC-independent manner to enrich H4K20me1 and facilitate chromosome compaction. Thus, DPY-21 is an adaptable chromatin regulator that is harnessed during development for distinct biological functions. In both somatic cells and germ cells, H4K20me1 enrichment modulates 3D chromosome architecture to carry out these functions.
Step of transcription controlled by the DCC.
We have dissected a key aspect of the dosage compensation mechanism by determining the step of transcription controlled by the DCC to repress X-chromosome gene expression. This work was performed in collaboration with John Lis' lab at Cornell University. In principle, the DCC could control any step of transcription: recruitment of RNA polymerase II (Pol II) to the promoter, initiation of transcription, escape of Pol II from the promoter or pause sites, elongation of RNA transcripts, or termination of transcription. The mechanism had been elusive in C. elegans due to improper annotation of transcription start sites (TSSs). Nascent RNA transcripts from most nematode genes undergo rapid co-transcriptional processing in which the 5' end is replaced by a common 22-nucleotide leader RNA through a trans-splicing mechanism, thereby destroying all knowledge of TSSs and promoters.
To understand the step of transcription controlled by the DCC, we first devised a general strategy for mapping transcription start sites and created an invaluable nematode TSS data set. The TSS mapping strategy, called GRO-cap, recovered nascent RNAs with 5'-caps prior to processing. We then determined the genome-wide distribution, orientation, and quantity of transcriptionally-engaged RNA Polymerase II (Pol II) relative to TSSs in wild-type and DC-defective animals using GRO-seq (global run-on sequencing).
We found that promoters are unexpectedly far upstream from the 5' ends of mature mRNAs, and promoter-proximal Pol II pausing occurs only in starved larvae and is rare in C. elegans embryos, unlike in most metazoans. These results indicated that enhancement of promoter pausing in XX embryos cannot be the mechanism of reducing transcription during dosage compensation. In contrast, control of pausing is a common mechanism for controlling transcription of developmental regulatory genes in most metazoans and is thought to be the mechanism of dosage compensation in fruit flies.
Then, by comparing the location and density of transcriptionally engaged Pol II in wild-type and dosage-compensation-defective embryos, we found that the step of transcription controlled by the dosage compensation process is the recruitment of Pol II. That is, C. elegans equalizes X-chromosome-wide gene expression between the sexes by reducing Pol II recruitment to the promoters of X-linked genes in XX embryos by about half. One of our research directions is to dissect the mechanisms by which the DCC limits Pol II recruitment.
Our data set also enabled us to analyze starvation-controlled gene regulation in collaboration with Ryan Baugh's lab at Duke University. We found a new phenomenon of Pol II docking, the stable association of Pol II upstream of the transcription start sites, and hence sites of pausing. We found that docked Pol II accumulates, without initiating, upstream of inactive growth genes that are turned off during starvation are activated upon feeding. We found that Pol II pausing occurs at active stress-response genes that are downregulated upon feeding. Hence, growth and stress genes are controlled by distinct mechanisms to coordinate gene expression with nutrient availability.
DCC recruitment and binding to X chromosomes.
We showed that many of the DCC recruitment (rex) sites have a DNA motif (called MEX) that is highly enriched on X compared to autosomes and is essential for DCC binding to a subset of rex sites. However, not all rex sites have this motif. We recently
defined new principles by which the DCC is recruited to X chromosomes, including the identification of a new, essential DCC binding motif (MEX II) that is enriched on X. We found that MEX II acts in combination with MEX to foster high-affinity binding at some rex sites but also acts alone at other rex sites to foster stable binding. We demonstrated these DCC binding principles by using DCC binding assays in vivo and in vitro.
We also showed that SUMOylation of specific DCC subunits is essential for sex-specific assembly and function of the DCC on X. Depletion of SUMO in vivo severely disrupts DCC binding and causes changes in X-linked gene expression similar to those caused by deleting the genes that encode DCC subunits. Three DCC subunits undergo SUMOylation, one subunit essential for DCC loading and two subunits that are integral to the condensin portion of the DCC.
DCC SUMOylation is triggered by the signal that initiates DCC assembly onto X. The initial step of assembly--binding of X-targeting factors to rex sites--is independent of SUMOylation, but robust binding of the complete complex requires SUMOylation. One of SUMOylated DCC subunits also participates in condensin complexes essential for chromosome segregation, but its SUMOylation occurs only in the context of the DCC. Our results reinforce a newly emerging theme in which multiple proteins of a complex are collectively SUMOylated in response to a specific stimulus, leading to accelerated complex formation and enhanced function.
Condensin-driven remodeling of X-chromosome topology during dosage compensation.
The three-dimensional organization of a genome plays a critical role in regulating gene expression, yet little is known about the machinery and mechanisms that determine higher-order chromosome structure. The involvement of bona fide condensin subunits in dosage compensation together with our observation that the DCC acts at a distance to regulate gene expression suggested that the DCC might alter the topology of X chromosomes to reduce gene expression chromosome wide.
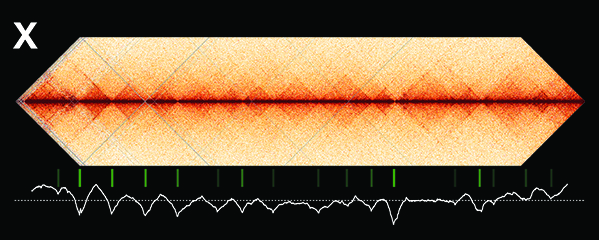
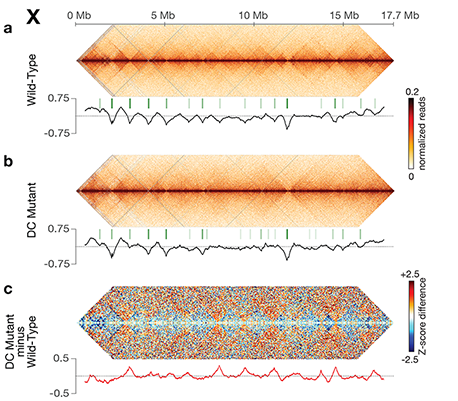
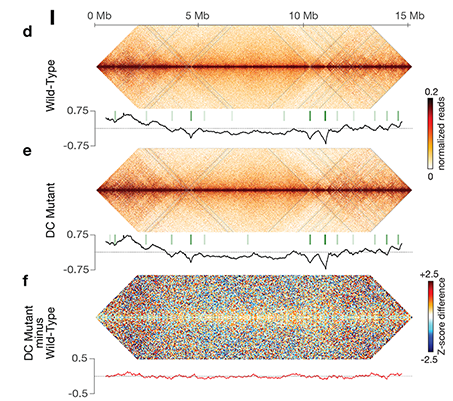
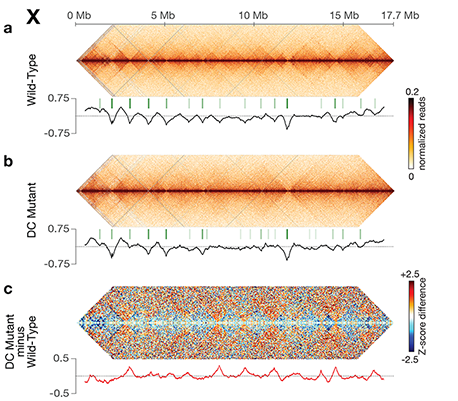
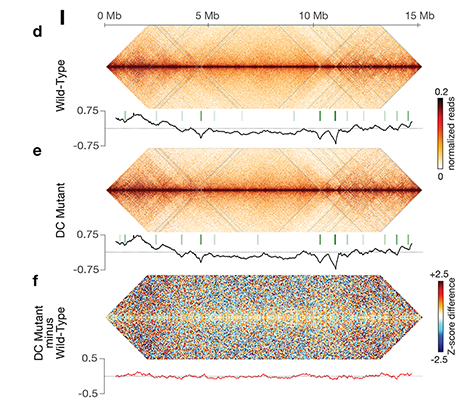
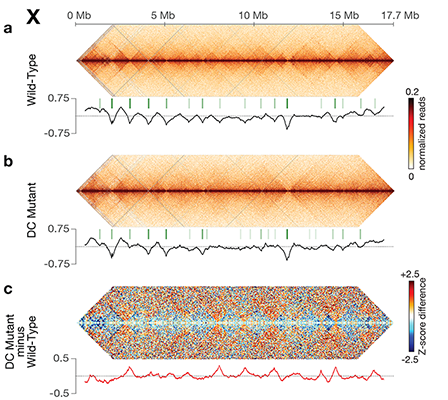
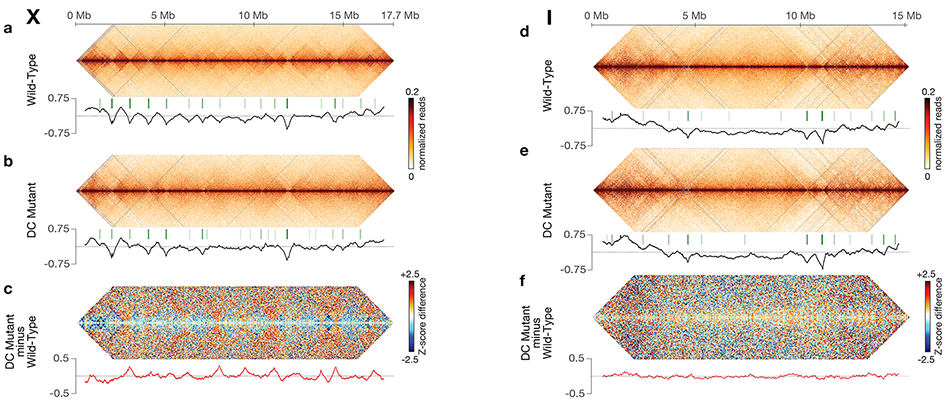
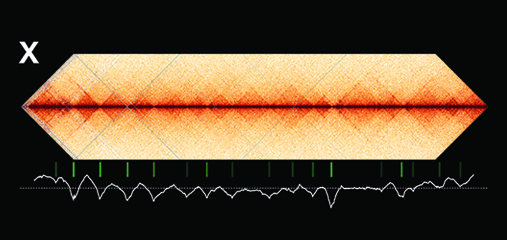
Using genome-wide chromosome conformation capture techniques (in collaboration with Job Dekker's lab at U. Mass. Worcester) with single-cell fluorescence in situ hybridization and RNA-seq to compare chromosome structure and gene expression in wild-type and dosage-compensation-defective embryos, we showed that the DCC remodels X chromosomes of hermaphrodites into a unique, sex-specific spatial conformation, distinct from autosomes, using its highest-affinity rex sites to facilitate long-range interactions across X. Dosage-compensated X chromosomes consist of self-interacting domains (~ 1 Mb) resembling mammalian Topologically Associating Domains (TADs). TADs on X have stronger boundaries and more regular spacing than those on autosomes. Many TAD boundaries on X coincide with the highest-affinity rex sites, and these boundaries become diminished or lost in mutants lacking DCC binding, causing the structure of X to resemble that of autosomes. These results predicted that deletion of an endogenous rex site at a DCC-dependent boundary should disrupt the boundary. As predicted, Cas9-mediated deletion of a rex site greatly diminished the boundary, further demonstrating the condensin-driven remodeling of X-chromosome topology during dosage compensation. Thus, condensin acts as a key structural element to reorganize interphase chromosomes and thereby regulate gene expression. Prior to our work, no molecular trigger or set of DNA binding sites was known to cause a comparably strong effect on TAD structure in higher eukaryotes. Our understanding of the topology of dosage-compensated X chromosomes provides fertile ground to decipher the detailed mechanistic relationship between higher-order chromosome structure and chromosome-wide regulation of gene expression.
X-Chromosome Domain Architecture Regulates C. elegans Lifespan but Not Dosage Compensation.
Interphase chromosomes are organized into a series of structures ranging from kilobase-scale chromatin loops to one megabase-scale topologically associating domains (TADs) and hundred-megabase territories. Mechanisms that establish these higher-order chromosome structures and their roles in gene regulation have been elusive.
Understanding the relationship between TAD structure and gene expression in mammalian cells has been challenging because architectural proteins that establish TADs also bind and function at locations other than TAD boundaries, such as promoters, making it unclear whether transcriptional changes resulting from their depletion are caused by altered TAD structure or by the proteins' other roles in gene regulation. Furthermore, the architectural proteins that establish mammalian TADs, such as condensin complexes, also play roles in essential cellular processes such as chromosome segregation, making the significance of TADs difficult to assess at the organismal level by depleting the proteins.
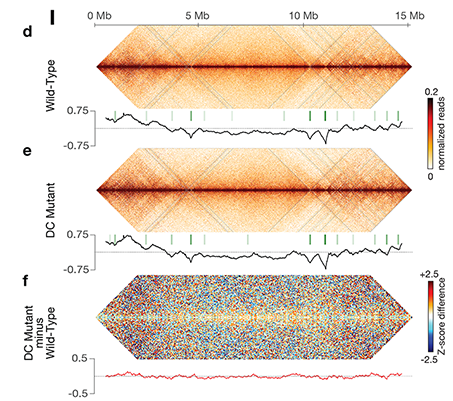
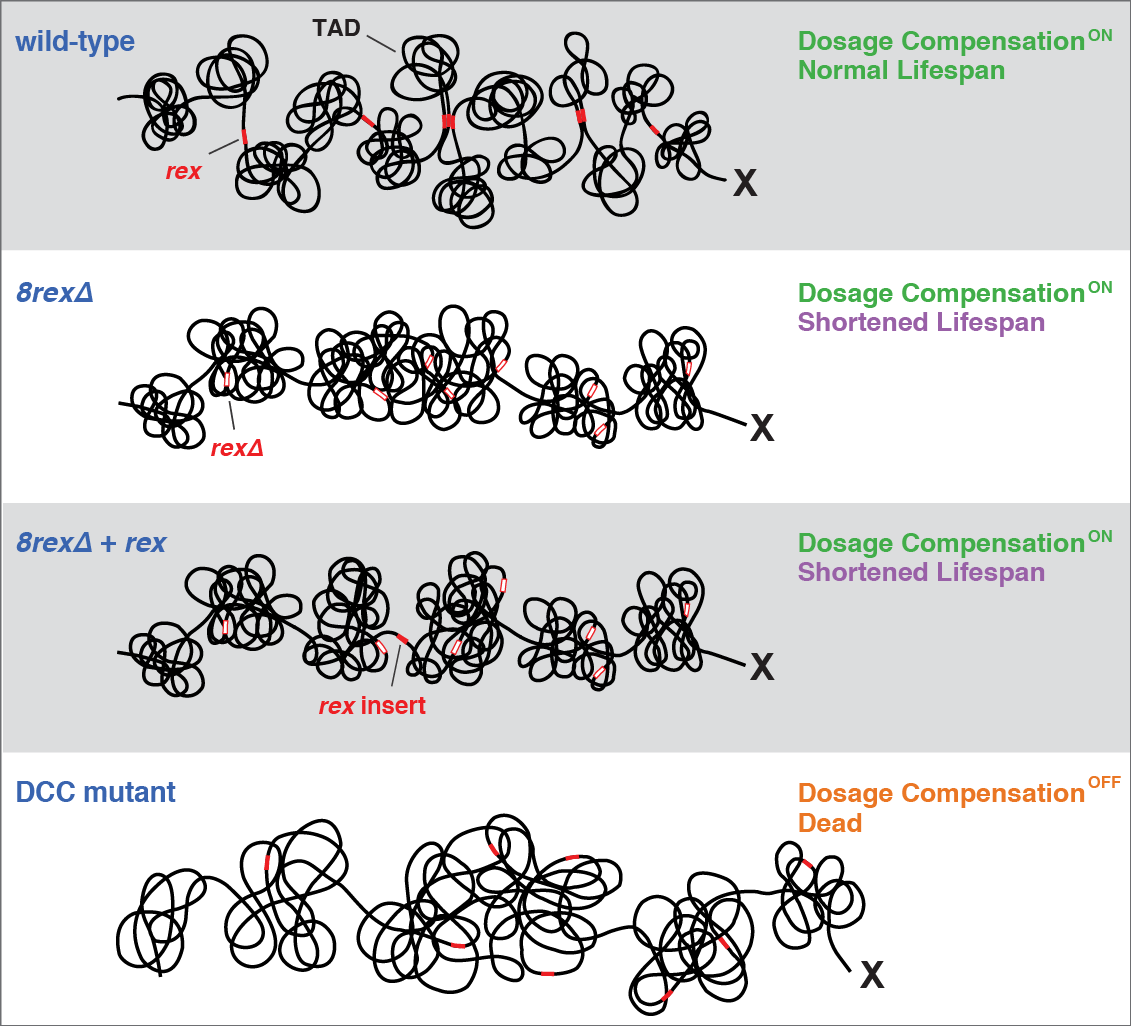
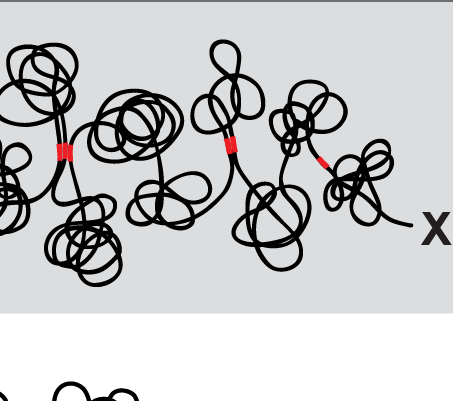
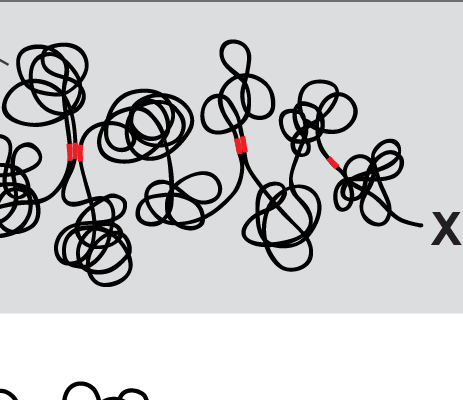
X chromosome dosage compensation in C. elegans has been ideal for dissecting the roles of TADs. Binding of the condensin DCC to X results in eight DCC-dependent TAD boundaries. All eight boundaries coincide with a high-affinity DCC rex site. Without DCC binding, the eight TAD boundaries are lost, causing X structure to resemble that of autosomes with fewer, less regularly spaced TAD boundaries. These remaining boundaries on X are DCC independent. Rather than depleting condensin subunits to disrupt TADs, we dissected the mechanism of TAD formation and the function of TADs by deleting a series of rex sites at TAD boundaries. We then measured the resulting chromosome structure and assessed the effect on gene expression and animal development. We also inserted high-affinity rex sites at new locations on 8rexΔ and wild-type X chromosomes to determine whether one rex site is sufficient to establish a new TAD boundary.
Each rex deletion eliminated the associated DCC-dependent TAD boundary, revealing that DCC binding at a high-occupancy rex site is necessary for boundary formation. Insertion of a rex site at a new location on X defined a new boundary, indicating that DCC binding at a high-occupancy rex site is sufficient to define a boundary on X. Deleting all eight rex sites at the eight DCC-dependent boundaries recapitulated the TAD structure of a DCC mutant. These 8rexΔ animals provided a unique opportunity to measure transcription when TAD structure was grossly disrupted across an entire metazoan chromosome but binding of the key architectural protein complex persisted on the numerous remaining rex sites. The 8rexΔ worms lacked canonical dosage compensation phenotypes and had normal compaction of X chromosomes. Embryos did not show statistically significant changes in X-chromosome expression, indicating that TAD structure does not drive dosage compensation. The absence of TADs allowed us to identify additional DCC-mediated X-chromosome structure: the DCC promotes DNA interactions across X between loci within 0.1-1 Mb. These TAD-independent interactions may underlie X compaction and be important for transcriptional repression. Although abrogating TAD structure in hermaphrodites by deleting rex sites did not disrupt dosage compensation, it did reduce thermotolerance, accelerate aging, and shorten lifespan, implicating chromosome architecture in stress responses and aging.
Targeted Genome-editing Across Highly Diverged Nematode Species.
Thwarted by the lack of reverse genetic approaches to enable cross-species comparisons of gene function, we established robust strategies for targeted genome editing across nematode species diverged by 300 MYR. In our initial work, a collaboration with Sangamo BioSciences, we used engineered nucleases containing fusions between the DNA cleavage domain of the enzyme FokI and a custom-designed DNA binding domain: either zinc-finger motifs for zinc-finger nucleases or transcription activator-like effector domains for TALE nucleases (TALENs). In those experiments, we allowed the DNA double-strand breaks to be repaired imprecisely by non-homologous end joining (NHEJ) to create mutations in precise locations.
We then extended the use of TALENs to achieve precise insertion and deletion of desired sequences by introducing single-stranded or double-stranded templates to generate precise insertions or deletions through homology directed repair (HDR), the first demonstration of HDR using ZFNs or TALENs in the nematode community. We then adopted the use of the CRISPR-associated nuclease Cas9 because of the ease in making RNA guides to program target specificity.
Despite successful application of Cas9 technology, predicting DNA targets and guide RNAs that support efficient genome editing was problematic. We then devised a strategy for high-frequency genome editing (both NHEJ and HDR) at all targets tested. The key innovation was designing guide RNAs with a GG motif at the 3' end of their target-specific sequences. This design increased the frequency of mutagenesis 10-fold. The ease of mutant recovery was further enhanced by combining this efficient guide design with a co-conversion strategy, in which targets of interest are analyzed in animals exhibiting a dominant phenotype caused by Cas9-dependent editing of an unrelated target.
Evolution cis-acting Regulatory Sites that Control Dosage Compensation.
Mechanisms that specify sexual fate and compensate for X-chromosome dose have diverged rapidly across species compared to other developmental processes, making it particularly informative to study these rapidly changing processes over short evolutionary time scales. Application of our genome editing strategies to C. briggsae revealed that the core dosage
compensation machinery and key components of the genetic hierarchy that controls dosage compensation and sex determination were conserved across the 30 MYR separation between C. elegans and C. briggsae. In contrast, the set of
cis-acting elements on X that recruit the DCC (rex sites) has diverged, retaining no functional overlap. ChIP-seq analysis defined the C. briggsae DCC binding sites, and in vivo binding assays confirmed the ability of these sites to recruit the DCC when
detached from X in C. briggsae but not in C. elegans, and vice versa. The evolution of these sites differs dramatically from the highly conserved DCC binding sites used by equivalently diverged fruit fly species and from the unchanged target sites of conserved transcription factors that control multiple developmental processes from flies to humans. Hence, the divergence in DCC binding specificity across nematode species provides a powerful opportunity to understand the path and timing for the concerted change in hundreds of DNA target sites and the evolution of X chromosomes. We have extended our analysis of DCC binding specificity to other nematode species and have shown that rex sites have diverged functionally at least three times in 30 MYR of evolutionary history.
Tethering Replicated Chromosomes via Cohesin to Ensure Genome Stability during Meiosis.
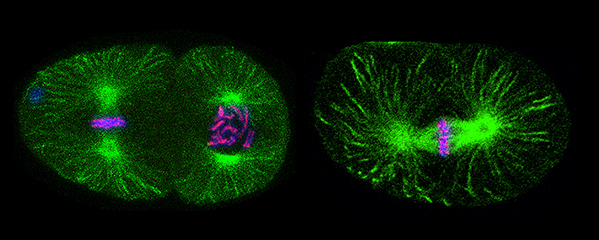
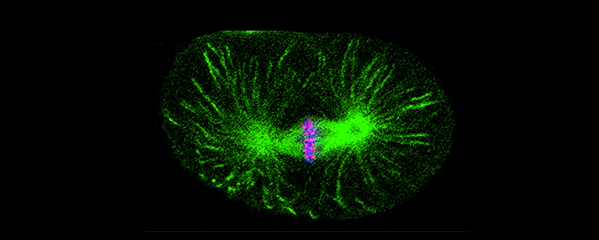
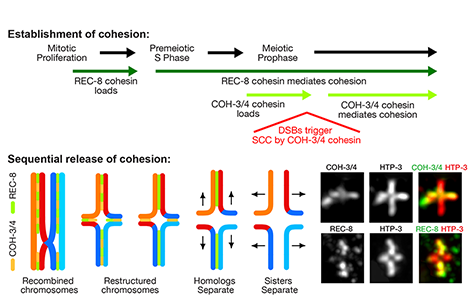
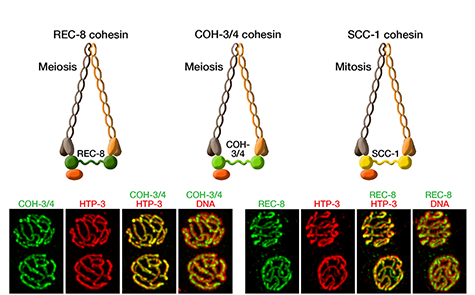
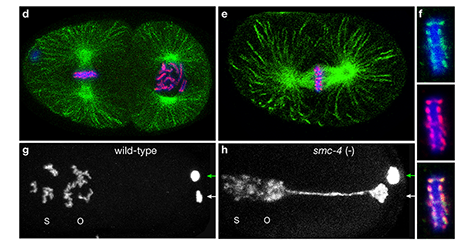
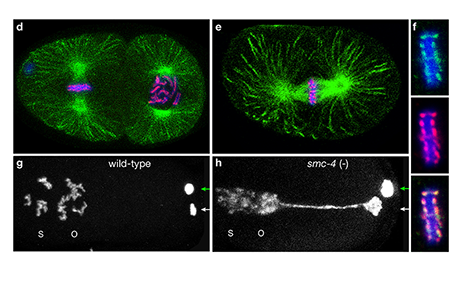
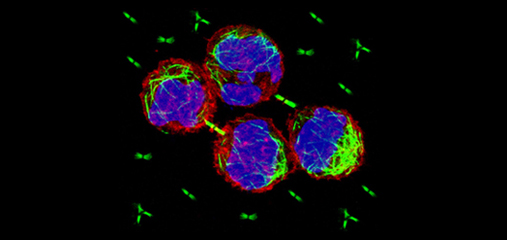
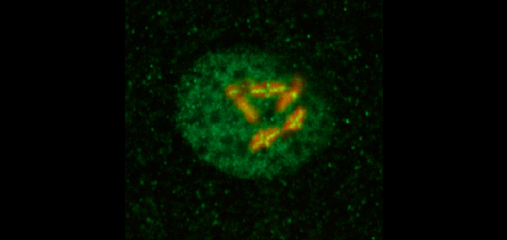
Faithful segregation of chromosomes during cell division is essential for genome stability. Accurate chromosome segregation is required both for the proliferative cell divisions that produce daughter cells during mitosis and the two sequential divisions that produce haploid sperm and eggs from diploid germline stem cells during meiosis. Approximately 30% of human zygotes have abnormal chromosome content at conception due to defects in meiosis. Such aneuploidy is a leading cause of miscarriages and birth defects and arises, in part, from defects in sister chromatid cohesion (SCC). SCC tethers replicated sister chromatids prior to cell divisions to ensure proper chromosome segregation. In humans, SCC is established in the developing germ cells of a fetus and must be maintained until ovulation in adults. This long-lived SCC is established and maintained by cohesin complexes, evolutionarily conserved protein complexes structurally related to condensin (Figure 7).
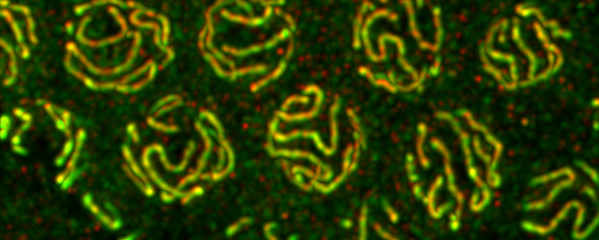
Studies in budding yeast showed that mitotic and meiotic cohesins are distinct but differ only in a single subunit called the kleisin. During yeast meiosis, a single cohesin complex carries out all aspects of SCC. In contrast, our work in nematodes shows that regulation of meiotic SCC in higher eukaryotes is more complex. We found that multiple functionally specialized cohesin complexes mediate the establishment and two-step release of SCC that underlies the production of haploid gametes (Figure 7). The meiotic complexes differ by a single kleisin subunit, and the kleisin influences nearly all aspects of meiotic cohesin function: the mechanisms for loading cohesins onto chromosomes, for triggering DNA-bound cohesins to become cohesive, and for releasing cohesins in a temporal- and location-specific manner (Figure 8). One kleisin triggers cohesion just after the chromosomes replicate, as in yeast. Unexpectedly, the other triggers cohesion in a replication-independent manner, only after programmed DSBs are made during meiosis to initiate recombination between homologous maternal and paternal chromosomes. Thus, break-induced cohesion is essential for tethering replicated meiotic chromosomes. Later, recombination stimulates separase-independent removal of the two different cohesin complexes from reciprocal chromosomal territories flanking the crossover site. This region-specific removal likely underlies the two-step separation of homologs and sisters. Unexpectedly, one cohesin complex also performs cohesion-independent functions in synaptonemal complex assembly. Our findings establish a new model for cohesin function in meiosis: the choreographed actions of multiple cohesins, endowed with unexpectedly specialized functions by their kleisins, underlie the stepwise separation of homologous chromosomes and then sister chromatids required for reduction of genome copy number. This model diverges significantly from that in yeast but likely applies to plants and mammals, which utilize similar meiotic kleisins.
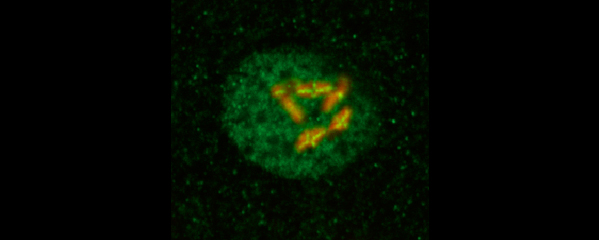
Meiotic Chromosome Structures Constrain and Respond to Designation of Crossover Sites.
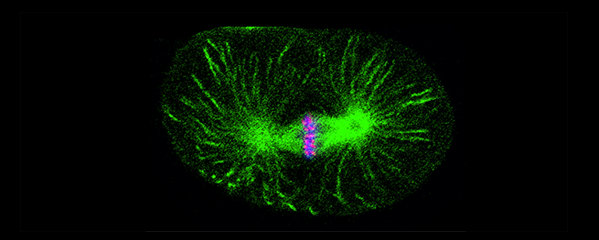
Crossover recombination events between homologous chromosomes are required to form chiasmata, temporary connections between homologues that ensure their proper segregation at meiosis I. Despite this requirement for crossovers and an excess of the double-strand DNA breaks that are the initiating events for meiotic recombination, most organisms make very few crossovers per chromosome pair. Moreover, crossovers tend to inhibit the formation of other crossovers nearby on the same chromosome pair, a poorly understood phenomenon known as crossover interference. We showed (in collaboration with the Villenueve lab at Stanford) that the synaptonemal complex, a meiosis-specific structure that assembles between aligned homologous chromosomes, both constrains and is altered by crossover recombination events. Partial depletion of the synaptonemal complex central region proteins attenuates crossover interference, increasing crossovers and reducing the effective distance over which interference operates, indicating that synaptonemal complex proteins limit crossovers. Moreover, we showed that crossovers are associated with a local 0.4-0.5-micrometre increase in chromosome axis length. We proposed that meiotic crossover regulation operates as a self-limiting system in which meiotic chromosome structures establish an environment that promotes crossover formation, which in turn alters chromosome structure to inhibit other crossovers at additional sites.
Ironically, the effect of depleting condensin I or condensin II on increasing crossovers appears to occur by a different mechanism, because the sites of extra crossovers are not marked by the same molecular markers as the crossovers created by reducing the synaptonemal complex. We are investigating the crossover pathway employed to achieve these extra, non-interfering crossovers in condensin mutants.